binnacle
1.0.0
К
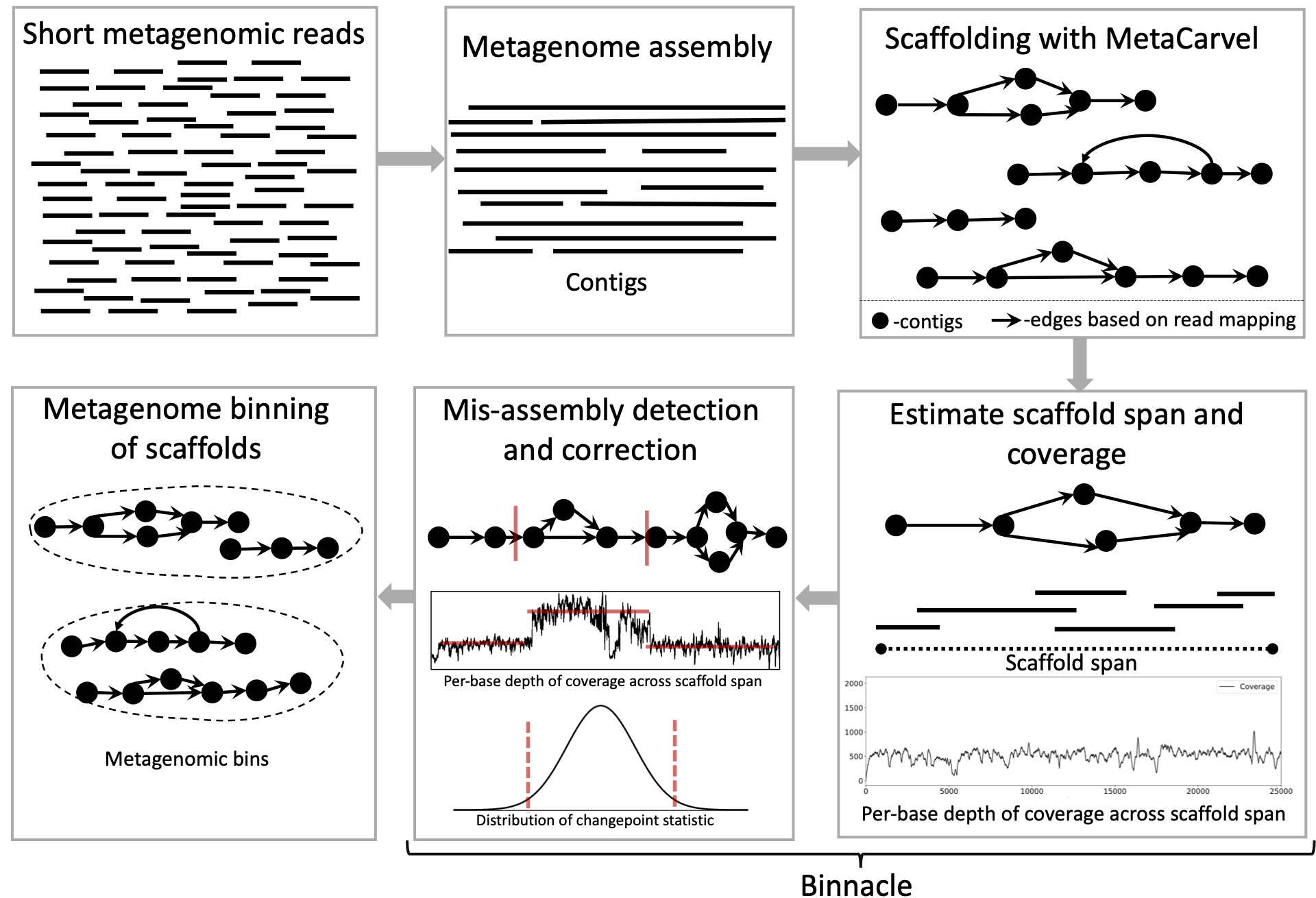
Binnacle точно вычисляет охват графических каркасов и плавно интегрируется с ведущими методами биннинга, такими как Metabat2, Maxbin 2.0 и Concoct. Использование графических каркасов, в отличие от контигов (наиболее распространенного подхода) для биннинга, улучшает смелость и качество метагеномных бункеров и может захватить более широкий набор вспомогательных элементов реконструированных геномов.
Чтобы запустить Binnacle, вам понадобятся Python 3.7.x, Bedtools, Samtools, Biopython, Networkx, Numpy и Pandas.
Доступен файл среды. Подробная документация о том, как установить эти пакеты, приведена здесь. Мы используем графические каркасы, которые являются выводами инструмента каркаса метакарвера, поэтому вам также необходимо будет загрузить и установить MetaCarvel. Существует пошаговое руководство по установке для MetaCarvel.
Как правило, когда у вас есть один или несколько метагеномных образцов, нам нужно собирать, карманы и бин -контиги/каркасы из каждого образца для генерации метагеномных бункеров. Мы рекомендуем использовать мегахит для сборки и метакарвеля для лесов. Мы предоставляем руководство для работы по работе с помощью сборки, лесов и шагов оценки охвата на базу.
Следуйте этим шагам, чтобы сгенерировать файлы для запуска методов биннинга с графическими каркасами:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Пожалуйста, проверьте вики для подробного описания на настройке среды Python, методах расчета охвата и типичного рабочего процесса для запуска Binnacle.
Чтобы визуализировать графические каркасы, мы рекомендуем использовать метагеномерзоп, который является веб-браузером. Вход в метагеномескоп - это adsembly_graph_filtered.gml. Подробная документация по установке и запуску метагеномескопа приведена здесь.
Пожалуйста, цитируйте Muralidharan HS, Shah N, Meisel JS и Pop M (2021) Binnacle: Использование каркасов для улучшения смежности и качества метагеномных бункеров. Передний. Микробиол. 12: 638561. doi: 10.3389/fmicb.2021.638561.
Этот инструмент все еще находится в стадии разработки. Пожалуйста, откройте проблему здесь, на GitHub, или свяжитесь с нами, если у вас есть какие -либо вопросы.
Harihara Muralidharan: [email protected]
Нидхи Шах: [email protected]


