ClockstaR
1.0.0

เซบาสเตียน ดูเชเน, มาร์ตีน่า โมลัก และไซมอน วายดับเบิลยู โฮ
ห้องปฏิบัติการนิเวศวิทยาระดับโมเลกุล วิวัฒนาการ และสายวิวัฒนาการ (MEEP)
โรงเรียนวิทยาศาสตร์ชีวภาพ
มหาวิทยาลัยซิดนีย์
10 มิถุนายน 2558
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
ใช้การปรับระยะห่างของต้นไม้ให้เหมาะสมโดยใช้อนุพันธ์ของระยะทาง BSD
ใช้เวอร์ชันขนานสำหรับระยะทางโทโพโลยี
เขียนบทช่วยสอนสำหรับการจัดกลุ่มระยะทางโทโพโลยี
รวม modelgenerator สำหรับการทดสอบโมเดล
ผสานรวม RaxML เพื่อเพิ่มความเป็นไปได้สูงสุดของความยาวสาขาและโทโพโลยี
การประมาณช่วงเวลาวิวัฒนาการด้วยชุดข้อมูลหลายยีนเป็นแบบฝึกหัดทั่วไปในการศึกษาสายวิวัฒนาการ ชุดข้อมูลหลายยีนสามารถแบ่งพาร์ติชันตามยีน ตำแหน่งโคดอน หรือทั้งสองอย่าง ในบทช่วยสอนนี้ เราเรียกว่า "ชุดย่อยของข้อมูล" ว่าเป็นยีนเดี่ยวๆ หรือหน่วยย่อยใดๆ ของชุดข้อมูลหลายยีน คำว่า “พาร์ติชัน” จะหมายถึงกลุ่มของชุดย่อยของข้อมูล
แม้ว่าชุดย่อยของข้อมูลสามารถต่อและวิเคราะห์ด้วยโมเดลนาฬิกาผ่อนคลายเดียว แต่รูปแบบของการเปลี่ยนแปลงอัตราระหว่างสายเลือดอาจแตกต่างกันระหว่างชุดย่อยของข้อมูล แม้ว่าโทโพโลยีแบบต้นไม้จะเหมือนกันก็ตาม ตัวอย่างเช่น ความแปรผันของอัตราระหว่างเชื้อสายในยีนไมโตคอนเดรียอาจแตกต่างจากยีนนิวเคลียร์ ดังนั้นจึงสามารถกำหนดแบบจำลองนาฬิกาผ่อนคลายที่แตกต่างกันให้กับชุดย่อยข้อมูลที่แตกต่างกันเพื่อปรับปรุงการประมาณค่าของช่วงเวลาวิวัฒนาการและความพอดีทางสถิติ (ดู Duchene และ Ho., 2014a)
มีหลายวิธีที่สามารถแบ่งพาร์ติชันชุดข้อมูลหลายยีนได้ วิธีการทั่วไปในการเปรียบเทียบโครงร่างการแบ่งพาร์ติชันคือการใช้ปัจจัย Bayes หรือเกณฑ์ตามความน่าจะเป็นสำหรับแบบจำลองที่พอดี อย่างไรก็ตาม ในกรณีส่วนใหญ่ เป็นไปไม่ได้ที่จะทดสอบรูปแบบการแบ่งพาร์ติชันที่เป็นไปได้ทั้งหมด โดยเฉพาะอย่างยิ่งกับวิธีการคำนวณที่เข้มข้นในการคำนวณปัจจัยเบย์
ClockstaR ประมาณความยาวสาขาสายวิวัฒนาการของแต่ละชุดข้อมูล ระยะทางของคะแนนสาขาหรือที่เรียกว่า sBSDmin ได้รับการคำนวณสำหรับต้นไม้ทุกคู่เพื่อใช้วัดความแตกต่างในรูปแบบความแปรผันของอัตราระหว่างสายเลือด ระยะทางเหล่านี้ใช้เพื่ออนุมานกลยุทธ์การแบ่งพาร์ติชันที่ดีที่สุดโดยใช้สถิติ GAP พร้อมอัลกอริธึมการจัดกลุ่ม PAM ตามที่นำมาใช้ในคลัสเตอร์แพ็คเกจ (Maechler et al., 2012) (สำหรับรายละเอียดของตัวชี้วัด sBSDmin ดู Duchene et al., 2014b) .
ClockstaR เป็นแพ็คเกจ R สำหรับการวิเคราะห์นาฬิกาโมเลกุลสายวิวัฒนาการของชุดข้อมูลหลายยีน ใช้รูปแบบของความแปรผันของอัตราเชื้อสายสำหรับยีนต่างๆ เพื่อเลือกกลยุทธ์การแบ่งพาร์ติชันนาฬิกา วิธีการนี้ใช้การวัดระยะทางจากต้นไม้สายวิวัฒนาการและอัลกอริธึมการเรียนรู้ของเครื่องที่ไม่ได้รับการดูแลเพื่อระบุจำนวนพาร์ติชั่นของนาฬิกาที่เหมาะสมที่สุด และยีนใดที่ควรวิเคราะห์ภายใต้แต่ละพาร์ติตอน สถานะการแบ่งพาร์ติชันที่เลือกใน ClocsktaR สามารถใช้สำหรับการวิเคราะห์นาฬิกาโมเลกุลในภายหลังด้วยโปรแกรมต่างๆ เช่น BEAST, MrBayes, PhyloBayes และอื่นๆ
โปรดไปที่ลิงก์นี้สำหรับสิ่งพิมพ์ต้นฉบับ
ClockstaR ต้องมีการติดตั้ง R นอกจากนี้ยังต้องมีการพึ่งพา R บางอย่างซึ่งสามารถรับผ่าน R ดังที่อธิบายไว้ด้านล่าง
กรุณาส่งคำขอหรือคำถามใดๆ ไปที่ Sebastian Duchene (sebastian.duchene[at]sydney.edu.au) ซอฟต์แวร์และทรัพยากรอื่นๆ สามารถพบได้ที่ห้องปฏิบัติการนิเวศวิทยาโมเลกุล วิวัฒนาการ และสายวิวัฒนาการแห่งมหาวิทยาลัยซิดนีย์
ดาวน์โหลดที่เก็บนี้เป็นไฟล์ zip แล้วแตกไฟล์ คำแนะนำต่อไปนี้ใช้โฟลเดอร์ clockstar_example_data ซึ่งมีไฟล์ fasta บางไฟล์และแผนผังสายวิวัฒนาการในรูปแบบ newick เปิดไฟล์เหล่านี้ในโปรแกรมแก้ไขข้อความ เช่น text wrangler ข้อมูลเหล่านี้ถูกจำลองภายใต้รูปแบบสี่รูปแบบของการแปรผันของอัตราการวิวัฒนาการ โปรดทราบว่าแผนผังคือโทโพโลยีแบบต้นไม้สำหรับยีนทั้งหมดหรือพาร์ติชันข้อมูล หากต้องการเรียกใช้ ClockstaR โปรดจัดรูปแบบข้อมูลของคุณให้คล้ายกับข้อมูลตัวอย่างใน clockstar_example_data
ClockstaR สามารถติดตั้งได้โดยตรงจาก GitHub สิ่งนี้ต้องการแพ็คเกจ devtools พิมพ์รหัสต่อไปนี้ที่พรอมต์ R เพื่อติดตั้งเครื่องมือที่จำเป็นทั้งหมด (โปรดทราบว่าคุณจะต้องเชื่อมต่ออินเทอร์เน็ตเพื่อดาวน์โหลดแพ็คเกจโดยตรง):
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )หลังจากดาวน์โหลดและติดตั้ง ให้โหลด ClockstaR ด้วย ไลบรารี ฟังก์ชัน
library (ClockstaR2)หากต้องการดูตัวอย่างเกี่ยวกับวิธีการรันโปรแกรม ให้พิมพ์:
example (ClockstaR2)ส่วนที่เหลือของบทช่วยสอนนี้ใช้โฟลเดอร์ clockstar_example_data
ขั้นตอนแรกคือการได้รับแผนผังยีนสำหรับการจัดตำแหน่งแต่ละตำแหน่ง ในการทำเช่นนี้ เราใช้โทโพโลยีแบบต้นไม้ และปรับความยาวกิ่งให้เหมาะสมโดยใช้การจัดตำแหน่งของยีนแต่ละรายการ ในกรณีนี้คือ A1.fasta ถึง C3.fasta หากคุณมีแผนผังยีน ให้บันทึกในรูปแบบ newick ในไฟล์และไปยังขั้นตอนถัดไป (เรียกใช้ clockstar แบบโต้ตอบ)
พิมพ์รหัสต่อไปนี้ในพรอมต์ R แล้วกด Enter:
optim . trees . interactive ()หากคุณได้รับข้อความแสดงข้อผิดพลาดเกี่ยวกับการติดตั้งแพ็คเกจ phangorn โปรดใช้โค้ดนี้แล้วทำซ้ำ optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR จะพิมพ์ข้อความต่อไปนี้:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICKลากโฟลเดอร์ clockstar_example_data ไปที่คอนโซล R แล้วพิมพ์ Enter โปรดทราบว่าโฟลเดอร์ควรมีเฉพาะการจัดเรียงในรูปแบบ FASTA และโทโพโลยีแบบต้นไม้ใน NEWICK คุณจะเห็นข้อความต่อไปนี้:
What should be the name of the file to save the optimised trees ?พิมพ์ชื่อไฟล์สำหรับแผนผังที่ปรับให้เหมาะสม ในกรณีนี้เราจะใช้ "example.trees"
example . treesณ จุดนี้ ClockstaR จะถามว่าควรใช้แบบจำลองการทดแทนแยกกันสำหรับแต่ละยีน หรือใช้ JC ในทุกกรณี เนื่องจากข้อมูลเหล่านี้จำลองภายใต้ JC เราจะพิมพ์ "n" แล้วกด Enter พิมพ์ "y" เพื่อระบุแต่ละรูปแบบการทดแทนแยกกัน
หลังจากพิมพ์ "n" แล้วกด Enter ClockstaR จะเริ่มทำงาน มันจะพิมพ์แผนผังยีนในอุปกรณ์กราฟิก หากต้นไม้ที่ระบุถูกรูทแล้ว ต้นไม้นั้นอาจพิมพ์คำเตือนบางอย่างด้วย ซึ่งสามารถเพิกเฉยได้อย่างปลอดภัย
เปิดโฟลเดอร์ clockstar_example_data คุณจะพบไฟล์ชื่อ "example.trees" ตามที่ระบุไว้ในขั้นตอนข้างต้น เปิด example.trees ในโปรแกรมแก้ไขข้อความ ประกอบด้วยแต่ละยีนทรีและชื่อต้นไม้ ตามชื่อของการจัดตำแหน่งของยีน มันควรมีลักษณะดังนี้:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.ไฟล์ที่มีต้นไม้นี้จะถูกใช้สำหรับขั้นตอนต่อไป
สำหรับขั้นตอนนี้ จำเป็นต้องมีแผนผังยีนในไฟล์ เช่นเดียวกับที่ได้รับในขั้นตอนก่อนหน้า
เปิด R และโหลด ClockstaR ตามที่แสดงด้านบน พิมพ์รหัสต่อไปนี้ที่พร้อมท์:
clockstar . interactive ()ClockstaR จะพิมพ์ข้อความต่อไปนี้:
please drag or type in the path to your gene trees file in NEWICK format :ลากไฟล์ที่มีแผนผังยีนไปยังคอนโซล R หากคุณทำตามขั้นตอนก่อนหน้านี้ ไฟล์จะถูกเรียกว่า example.trees พิมพ์ ป้อน
ClockstaR อาจถามว่าควรทำงานแบบขนานหรือไม่ ทั้งนี้ขึ้นอยู่กับแพ็คเกจที่คุณติดตั้ง สิ่งนี้มีประสิทธิภาพสำหรับชุดข้อมูลขนาดใหญ่ แต่สำหรับข้อมูลตัวอย่างจะไม่สร้างความแตกต่างมากนัก ดังนั้นให้พิมพ์ "n" หากคุณเห็นข้อความนี้แล้วพิมพ์ enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar จะเริ่มทำงานแล้ว ผลลัพธ์บนหน้าจอควรมีลักษณะดังนี้:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.หลังจากประมาณระยะทางของต้นไม้ (อธิบายไว้ในสิ่งพิมพ์ต้นฉบับ) ClockstaR จะพิมพ์ข้อความต่อไปนี้:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)นี่คือการตั้งค่าสำหรับอัลกอริธึมการจัดกลุ่ม เหมาะสำหรับชุดข้อมูลส่วนใหญ่ ดังนั้นในตัวอย่างนี้ เราสามารถพิมพ์ "y" แล้วป้อนได้ โดยการพิมพ์ "n" เราจะสามารถเปลี่ยนการตั้งค่าเหล่านี้ สำหรับรายละเอียดเพิ่มเติม โปรดดูที่ Kaufman และ Rousseeuw (2009)
ClockstaR จะเรียกใช้อัลกอริธึมการจัดกลุ่ม ในตอนท้ายมันจะพิมพ์พาร์ติชันจำนวนที่ดีที่สุดและถามว่าควรบันทึกผลลัพธ์เป็นไฟล์ pdf หรือไม่:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)พิมพ์ "y" จากนั้นป้อน
ClockstaR จะถามชื่อของไฟล์ที่ส่งออก:
What should be the name and path of the output file ?สำหรับตัวอย่างนี้ ให้พิมพ์ "example_run" แล้วป้อน แต่สามารถใช้ชื่อใดก็ได้
ตอนนี้เปิดโฟลเดอร์ clockstar_example_data แล้วเปิดไฟล์ pdf สองไฟล์ example_run_gapstats.pdf และ example_run_matrix.pdf
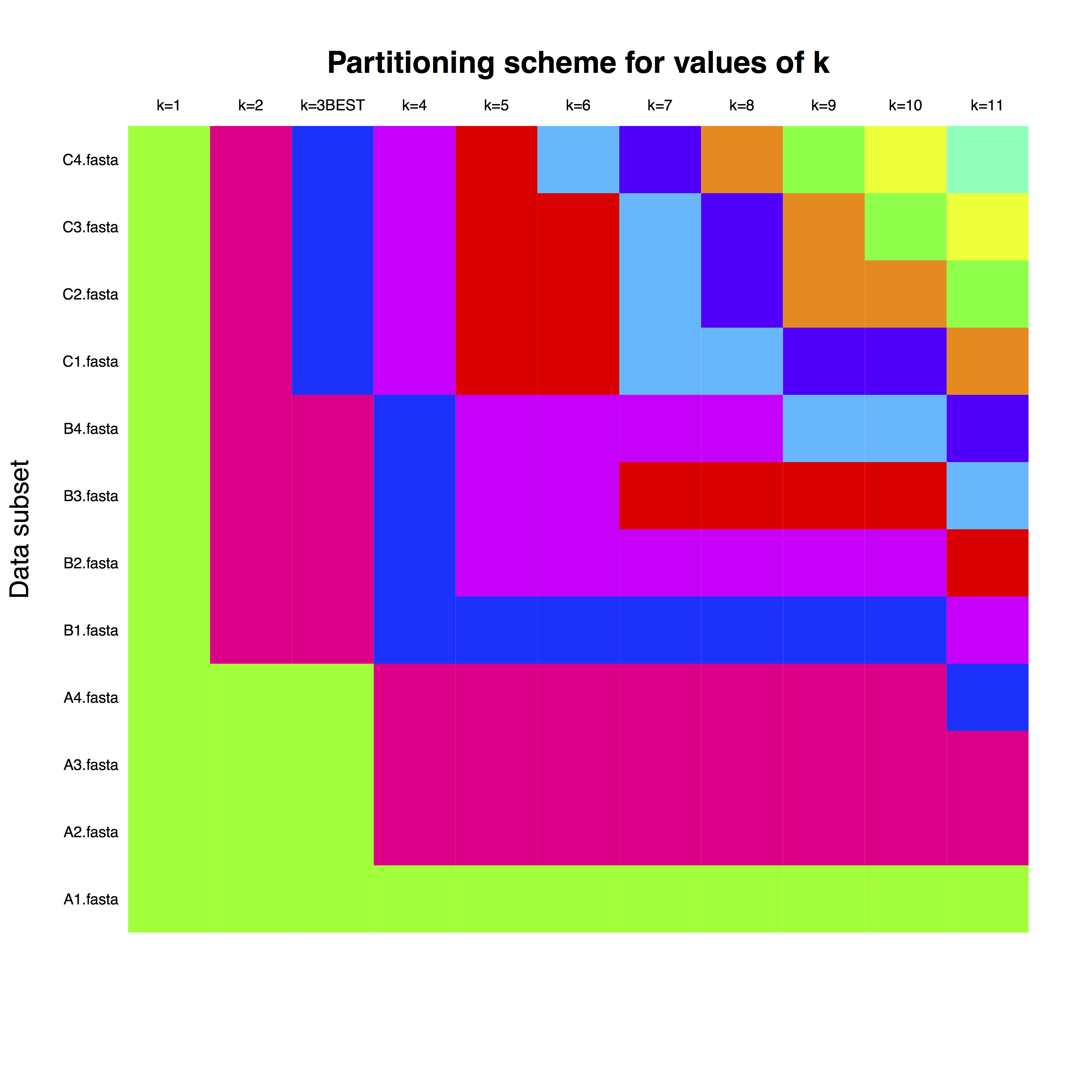
example_run_matrix คือเมทริกซ์ โดยที่แถวสอดคล้องกับแต่ละยีน ตามชื่อในไฟล์ FASTA คอลัมน์คือจำนวนพาร์ติชัน และสีแสดงถึงการกำหนดของแต่ละยีนให้กับพาร์ติชันนาฬิกา ตัวอย่างเช่น สำหรับ k =3 ซึ่งเป็นจำนวนพาร์ติชั่นที่ดีที่สุด เราสามารถใช้พาร์ติชั่นนาฬิกาแยกกันสำหรับยีนที่มีตัวอักษร A, B และ C
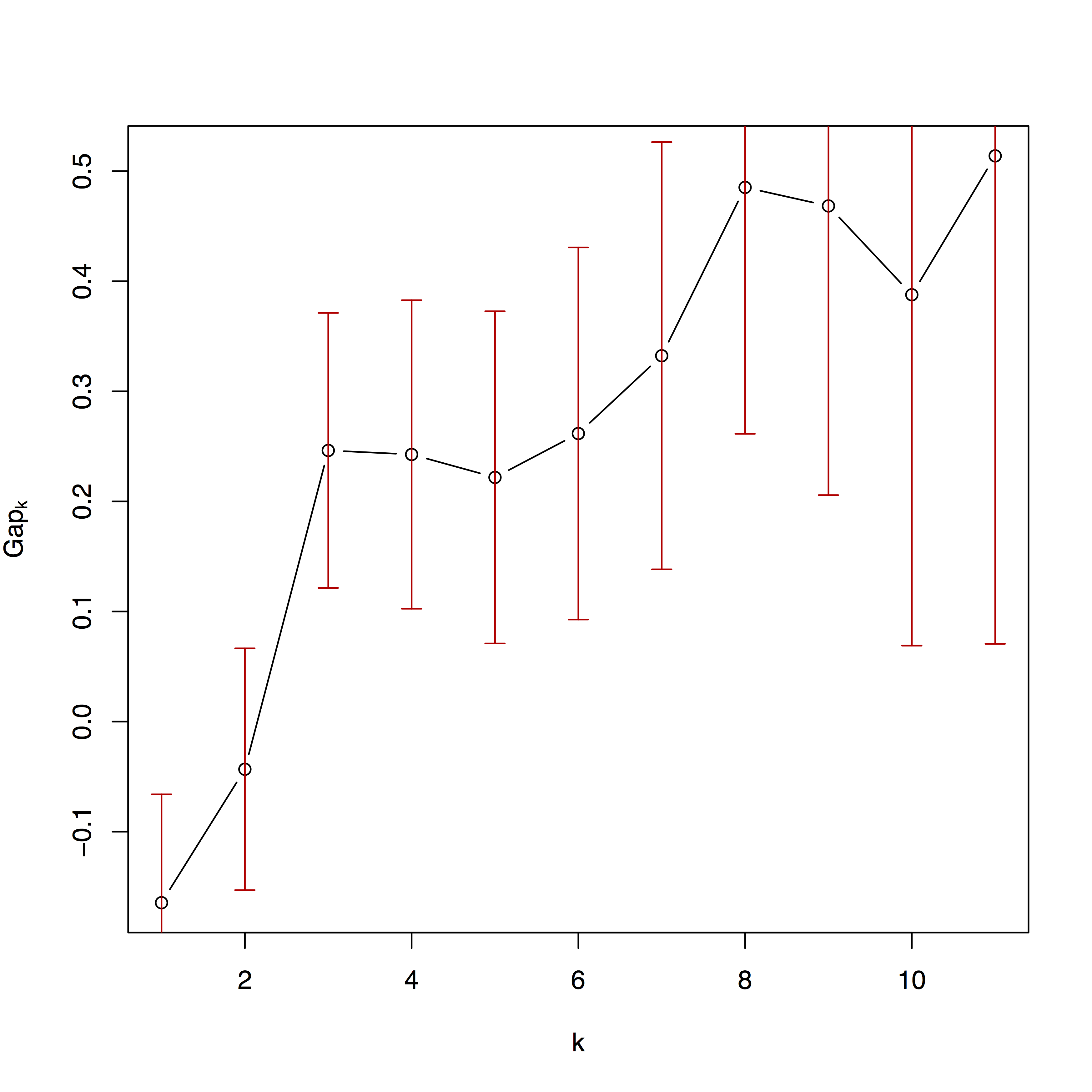
พล็อตที่สองคือความพอดีของอัลกอริธึมการจัดกลุ่มในพาร์ติชันจำนวนต่างๆ รายละเอียดเพิ่มเติมมีอยู่ใน Kaufman และ Rousseeuw (2009) และในเอกสารประกอบสำหรับคลัสเตอร์แพ็คเกจ
ClockstaR สามารถรันได้ด้วยการตั้งค่าแบบกำหนดเองอื่นๆ โปรดดูเอกสารประกอบสำหรับรายละเอียดอื่นๆ หรือส่งอีเมลมาหาฉันหากมีคำถามใดๆ ที่ sebastian.duchene[at]sydney.edy.au
โลโก้ได้รับการออกแบบโดย Jun Tong
ดูเชเน เอส. และโฮ SY (2014a) การใช้แบบจำลองนาฬิกาผ่อนคลายหลายแบบเพื่อประเมินช่วงเวลาวิวัฒนาการจากข้อมูลลำดับดีเอ็นเอ สายวิวัฒนาการระดับโมเลกุลและวิวัฒนาการ (77): 65-70
Duchene, S. , Molak, M. , & Ho, SY (2014b) ClockstaR: การเลือกจำนวนแบบจำลองนาฬิกาผ่อนคลายในการวิเคราะห์สายวิวัฒนาการระดับโมเลกุล ชีวสารสนเทศศาสตร์ 30(7): 1017-1019.
ลิตร, แอล. และรุสซีว, พีเจ (2009) การค้นหากลุ่มในข้อมูล: บทนำเกี่ยวกับการวิเคราะห์คลัสเตอร์ (เล่ม 344) จอห์น ไวลีย์ แอนด์ ซันส์


