reference_database_creator
bug fix --in-silico-pcr --untrimmed
ปู ( ค การรับประทานอาหาร ร ฐานข้อมูลอ้างอิงสำหรับ ก mplicon- บี ased ส equencing) เป็นโปรแกรมซอฟต์แวร์อเนกประสงค์ที่สร้างฐานข้อมูลอ้างอิงที่รวบรวมไว้สำหรับการวิเคราะห์เมเทเจโนมิก ขั้นตอนการทำงานของ CRABS ประกอบด้วยเจ็ดโมดูล: (i) ดาวน์โหลดข้อมูลจากที่เก็บข้อมูลออนไลน์; (ii) นำเข้าข้อมูลที่ดาวน์โหลดมาในรูปแบบ CRABS (iii) แยกบริเวณแอมพลิคอนผ่านการวิเคราะห์ ซิลิโก PCR (iv) ดึงข้อมูลแอมพลิคอนโดยไม่มีบริเวณที่มีผลผูกพันกับไพรเมอร์ผ่านการจัดตำแหน่งด้วยบาร์โค้ดที่สกัด ด้วยซิลิโก (v) ดูแลจัดการและย่อยฐานข้อมูลท้องถิ่นผ่านพารามิเตอร์การกรองหลายตัว (vi) ส่งออกฐานข้อมูลท้องถิ่นในรูปแบบต่าง ๆ ตามข้อกำหนดตัวแยกประเภทอนุกรมวิธาน และ (vi) ฟังก์ชั่นหลังการประมวลผล เช่น การสร้างภาพข้อมูล เพื่อสำรวจและให้ภาพรวมโดยสรุปของฐานข้อมูลอ้างอิงในพื้นที่ โมดูลทั้งเจ็ดนี้แบ่งออกเป็นสิบแปดฟังก์ชันและอธิบายไว้ด้านล่าง นอกจากนี้ ยังมีการจัดเตรียมโค้ดตัวอย่างสำหรับแต่ละฟังก์ชันทั้ง 18 ฟังก์ชัน สุดท้ายนี้ บทช่วยสอนเพื่อสร้างฐานข้อมูลอ้างอิงปลาฉลามในพื้นที่สำหรับชุดไพรเมอร์ MiFish-E มีให้ที่ส่วนท้ายของเอกสาร README นี้เพื่อให้มีสคริปต์ตัวอย่างสำหรับการอ้างอิง
เรารู้สึกตื่นเต้นที่จะประกาศว่า CRABS ได้เห็นการอัปเดตที่สำคัญและการออกแบบโค้ดใหม่ตามคำติชมของผู้ใช้ ซึ่งเราหวังว่าจะปรับปรุงประสบการณ์ผู้ใช้ในการสร้างฐานข้อมูลอ้างอิงในพื้นที่ของคุณเอง!
โปรดดูรายการคุณสมบัติและการปรับปรุงที่เพิ่มใน CRABS v 1.0.0 ด้านล่าง:
ขณะนี้ CRABS v 1.0.0 สามารถดาวน์โหลดได้ด้วยตนเองโดยการโคลนพื้นที่เก็บข้อมูล GitHub นี้ (ดู 4.1 การติดตั้งด้วยตนเอง สำหรับข้อมูลโดยละเอียด) เราจะอัปเดตคอนเทนเนอร์ Docker และแพ็คเกจ conda โดยเร็วที่สุดเพื่อให้ง่ายต่อการติดตั้งเวอร์ชันใหม่ล่าสุด
เมื่อใช้ CRABS ในโครงการวิจัยของคุณ โปรดอ้างอิงเอกสารต่อไปนี้:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS เป็นชุดเครื่องมือบรรทัดคำสั่งเท่านั้นที่ทำงานบนสภาพแวดล้อม Unix/Linux ทั่วไป และเขียนด้วยภาษา python3 โดยเฉพาะ อย่างไรก็ตาม CRABS ใช้โมดูล กระบวนการย่อย ใน python เพื่อรันคำสั่งหลายคำสั่งในรูปแบบ bash เพื่อหลีกเลี่ยงลักษณะเฉพาะของ python และเพิ่มความเร็วในการดำเนินการ เรามีสามวิธีในการติดตั้ง CRABS สำหรับ CRABS เวอร์ชันล่าสุด เราขอแนะนำให้ติดตั้งด้วยตนเองโดยการโคลนพื้นที่เก็บข้อมูล GitHub นี้และติดตั้งการขึ้นต่อกัน 10 รายการแยกกัน (คำแนะนำในการติดตั้งสำหรับการขึ้นต่อกันทั้งหมดที่มีให้ใน 4.1 การติดตั้งด้วยตนเอง) CRABS ยังสามารถติดตั้งผ่าน Docker และ conda ทั้งสองวิธีช่วยให้ติดตั้งได้ง่ายโดยติดตั้งการขึ้นต่อกันทั้งหมดร่วมกันโดยอัตโนมัติ เรามุ่งหวังที่จะรักษาคอนเทนเนอร์ Docker และแพ็คเกจ conda ให้ทันสมัยอยู่เสมอ แม้ว่าการอัปเดตเป็นเวอร์ชันใหม่ล่าสุดอาจเกิดความล่าช้าบ้าง โดยเฉพาะอย่างยิ่งสำหรับแพ็คเกจ conda ด้านล่างนี้เป็นรายละเอียดสำหรับทั้งสามแนวทาง
สำหรับการติดตั้งด้วยตนเอง ให้โคลนที่เก็บ CRABS ก่อน ขั้นตอนนี้ต้องการให้ GitHub พร้อมใช้งานกับบรรทัดคำสั่ง (คำแนะนำในการติดตั้งสำหรับ GitHub)
git clone https://github.com/gjeunen/reference_database_creator.git
อาจจำเป็นต้องทำให้ CRABS สามารถเรียกใช้งานได้บนระบบของคุณ ทั้งนี้ขึ้นอยู่กับการตั้งค่าของคุณ สามารถทำได้โดยใช้รหัสด้านล่าง
chmod +x reference_database_creator/crabs
เมื่อติดตั้ง CRABS แล้ว เราจำเป็นต้องตรวจสอบให้แน่ใจว่ามีการติดตั้งการขึ้นต่อกันทั้งหมดและสามารถเข้าถึงได้จากทั่วโลก CRABS เวอร์ชันล่าสุด (เวอร์ชัน v 1.0.0 ) ทำงานบน Python 3.11.7 (หรือเวอร์ชันที่เข้ากันได้กับ 3.11.7) และใช้โมดูล Python ห้าโมดูลที่อาจไม่ได้มาตรฐานกับ Python รวมถึงโปรแกรมซอฟต์แวร์ภายนอกห้าโปรแกรม การขึ้นต่อกันทั้งหมดแสดงไว้ด้านล่าง พร้อมด้วยลิงก์ไปยังคำแนะนำในการติดตั้ง หมายเลขเวอร์ชันที่ให้ไว้สำหรับแต่ละโมดูลและโปรแกรมซอฟต์แวร์คือหมายเลขที่ CRABS ได้รับการพัฒนา แม้ว่าแต่ละเวอร์ชันที่เข้ากันได้ก็สามารถใช้ได้เช่นกัน
โมดูลหลาม:
โปรแกรมซอฟต์แวร์ภายนอก:
เมื่อติดตั้ง CRABS และการอ้างอิงทั้งหมดแล้ว CRABS จะสามารถเข้าถึงได้ทั่วทั้งระบบปฏิบัติการโดยใช้โค้ดด้านล่าง
export PATH="/path/to/crabs/folder:$PATH"
แทนที่ /path/to/crabs/folder ด้วยพาธจริงไปยังโฟลเดอร์ที่เก็บ GitHub บนระบบปฏิบัติการ เช่น โฟลเดอร์ที่สร้างขึ้นระหว่างคำสั่ง git clone ด้านบน การเพิ่มโค้ด export ลงในไฟล์ .bash_profile หรือ .bashrc จะทำให้ CRABS สามารถเข้าถึงได้ทั่วโลกตลอดเวลา
Docker เป็นโครงการโอเพ่นซอร์สที่อนุญาตให้ปรับใช้แอปพลิเคชันซอฟต์แวร์ภายใน 'คอนเทนเนอร์' ที่แยกจากคอมพิวเตอร์ของคุณและทำงานผ่านระบบปฏิบัติการโฮสต์เสมือนที่เรียกว่า Docker Engine ข้อได้เปรียบหลักของการรันนักเทียบท่าบนเครื่องเสมือนคือใช้ทรัพยากรน้อยกว่ามาก การแยกส่วนนี้หมายความว่าคุณสามารถเรียกใช้คอนเทนเนอร์ Docker บนระบบปฏิบัติการส่วนใหญ่ รวมถึง Mac, Windows และ Linux คุณอาจต้องตั้งค่าบัญชีฟรีเพื่อใช้ Docker Desktop ลิงค์ นี้มีคำแนะนำที่ดีเกี่ยวกับพื้นฐานของการใช้ Docker นี่ คือลิงค์สำหรับการเริ่มต้นและมุ่งเน้นไปที่ Docker multiverse
มีเพียงสองขั้นตอนในการทำให้ Crabs ทำงานบนคอมพิวเตอร์ของคุณ ขั้นแรก ติดตั้ง Docker Desktop บนคอมพิวเตอร์ของคุณ ซึ่งฟรีสำหรับผู้ใช้ส่วนใหญ่ คำแนะนำสำหรับ Mac มีดังนี้ นี่คือคำแนะนำสำหรับคอมพิวเตอร์ Windows และ นี่คือคำแนะนำสำหรับ Linux (รองรับแพลตฟอร์ม Linux หลักๆ ส่วนใหญ่) เมื่อคุณติดตั้งและใช้งาน Docker Desktop แล้ว (ต้องเรียกใช้แอปพลิเคชันเดสก์ท็อปเพื่อให้คุณใช้คำสั่งนักเทียบท่าบนบรรทัดคำสั่ง) คุณเพียงแค่ต้อง 'ดึง' อิมเมจ Crabs ของเรา และคุณก็พร้อมที่จะไป:
docker pull quay.io/swordfish/crabs:0.1.7
แม้ว่าการติดตั้งแอปพลิเคชันนักเทียบท่าจะเป็นเรื่องง่าย แต่การใช้แอปพลิเคชันเหล่านั้นอาจยุ่งยากเล็กน้อยในช่วงแรก เพื่อช่วยคุณในการเริ่มต้น เราได้จัดเตรียมคำสั่งตัวอย่างโดยใช้ Crab เวอร์ชันนักเทียบท่า ตัวอย่างเหล่านี้สามารถพบได้ในโฟลเดอร์ docker_intro บน repo นี้ จากตัวอย่างเหล่านี้ คุณควรจะสามารถดำเนินการตั้งค่าฐานข้อมูลอ้างอิงทั้งหมดได้และพร้อมที่จะดำเนินการ เราจะขยายตัวอย่างเหล่านี้ต่อไปและทดสอบสิ่งนี้ในสถานการณ์ต่างๆ มากมาย โปรดถามคำถามและให้ข้อเสนอแนะในแท็บปัญหา
หากต้องการติดตั้งแพ็คเกจ conda คุณต้องติดตั้ง conda ก่อน ดูลิงค์นี้สำหรับรายละเอียด หากติดตั้ง conda ไว้แล้ว แนวทางปฏิบัติที่ดีในการอัพเดตเครื่องมือ conda ด้วย conda update conda ก่อนที่จะติดตั้ง CRABS
เมื่อติดตั้ง conda แล้ว ให้ทำตามขั้นตอนด้านล่างเพื่อติดตั้ง CRABS และการอ้างอิงทั้งหมด ตรวจสอบให้แน่ใจว่าได้ป้อนคำสั่งตามลำดับที่ปรากฏด้านล่าง
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
เมื่อคุณป้อนคำสั่งติดตั้งแล้ว conda จะประมวลผลคำขอ (อาจใช้เวลาประมาณหนึ่งนาที) จากนั้นจึงแสดงแพ็คเกจและโปรแกรมทั้งหมดที่จะติดตั้ง และขอให้ยืนยัน พิมพ์ y เพื่อเริ่มการติดตั้ง หลังจากเสร็จสิ้นขั้นตอนนี้ CRABS ควรจะพร้อมที่จะไป
เราได้ทดสอบการติดตั้งนี้บนระบบ Mac และ Linux เรายังไม่ได้ทดสอบบนระบบย่อย Windows สำหรับ Linux (WSL)
ใช้รหัสด้านล่างเพื่อตรวจสอบว่า CRABS ได้รับการติดตั้งสำเร็จหรือไม่ และดึงข้อมูลวิธีใช้ขึ้นมา
crabs -hข้อมูลวิธีใช้แบ่งฟังก์ชันทั้ง 18 ฟังก์ชันออกเป็นกลุ่มต่างๆ โดยแต่ละกลุ่มจะแสดงฟังก์ชันที่ด้านบนสุดและพารามิเตอร์ที่จำเป็นและเป็นทางเลือกด้านล่าง

CRABS ประกอบด้วยเจ็ดโมดูลซึ่งรวมเอาฟังก์ชันสิบแปดฟังก์ชัน:
โมดูล 1: ดาวน์โหลดข้อมูลจากที่เก็บข้อมูลออนไลน์
--download-taxonomy : ดาวน์โหลดข้อมูลอนุกรมวิธานของ NCBI;--download-bold : ดาวน์โหลดข้อมูลลำดับจากฐานข้อมูลบาร์โค้ดแห่งชีวิต (BOLD);--download-embl : ข้อมูลลำดับการดาวน์โหลดจาก European Nucleotide Archive (ENA; EMBL);--download-mitofish : ดาวน์โหลดข้อมูลลำดับจากฐานข้อมูล MitoFish--download-ncbi : ข้อมูลลำดับการดาวน์โหลดจากศูนย์ข้อมูลเทคโนโลยีชีวภาพแห่งชาติ (NCBI)โมดูล 2: นำเข้าข้อมูลที่ดาวน์โหลดเป็นรูปแบบ CRABS
--import : นำเข้าลำดับที่ดาวน์โหลดหรือบาร์โค้ดที่กำหนดเองเป็นรูปแบบ CRABS--merge : รวมไฟล์ที่จัดรูปแบบ CRABS ต่างๆ ให้เป็นไฟล์เดียวโมดูล 3: แยกบริเวณแอมพลิคอนผ่านการวิเคราะห์ ด้วยซิลิโก PCR
--in-silico-pcr : แยกแอมพลิคอนจากข้อมูลที่ดาวน์โหลดโดยการค้นหาและลบขอบเขตที่มีผลผูกพันกับไพรเมอร์โมดูล 4: ดึงข้อมูลแอมพลิคอนโดยไม่มีบริเวณที่มีผลผูกพันกับไพรเมอร์
--pairwise-global-alignment : ดึงข้อมูลแอมพลิคอนโดยไม่มีขอบเขตที่มีผลผูกพันกับไพรเมอร์โดยการจัดลำดับที่ดาวน์โหลดให้ตรงกับบาร์โค้ดที่แยกออกมา ในซิลิโกโมดูล 5: ดูแลจัดการและย่อยฐานข้อมูลท้องถิ่นผ่านพารามิเตอร์การกรองหลายตัว
--dereplicate : ละทิ้งลำดับที่ซ้ำกัน;--filter : ละทิ้งลำดับผ่านพารามิเตอร์การกรองหลายตัว--subset : ย่อยฐานข้อมูลท้องถิ่นเพื่อรักษาหรือแยกกลุ่มอนุกรมวิธานที่ระบุโมดูล 6: ส่งออกฐานข้อมูลท้องถิ่น
--export : ส่งออกฐานข้อมูลที่จัดรูปแบบ CRABS เป็นรูปแบบต่างๆ ตามข้อกำหนดของตัวแยกประเภทอนุกรมวิธานที่จะใช้โมดูล 7: ฟังก์ชันหลังการประมวลผลเพื่อสำรวจและให้ภาพรวมโดยสรุปของฐานข้อมูลอ้างอิงในเครื่อง
--diversity-figure : สร้างแผนภูมิแท่งแนวนอนที่แสดงจำนวนชนิดและกลุ่มลำดับต่อระดับที่ระบุรวมอยู่ในฐานข้อมูลอ้างอิง--amplicon-length-figure : สร้างแผนภูมิเส้นที่แสดงการแจกแจงความยาวของแอมพลิคอนแยกจากกลุ่มอนุกรมวิธาน--phylogenetic-tree : สร้างต้นไม้สายวิวัฒนาการด้วยบาร์โค้ดจากฐานข้อมูลอ้างอิงสำหรับรายการเป้าหมายของสายพันธุ์--amplification-efficiency-figure : สร้างกราฟแท่งที่แสดงความไม่ตรงกันในภูมิภาคที่มีผลผูกพันกับไพรเมอร์--completeness-table : สร้างสเปรดชีตที่มีความพร้อมใช้งานของบาร์โค้ดสำหรับกลุ่มอนุกรมวิธานCRABS สามารถดาวน์โหลดข้อมูลลำดับเริ่มต้นได้จากที่เก็บข้อมูลออนไลน์สี่แห่ง รวมถึง (i) BOLD, (ii) EMBL, (iii) MitoFish และ NCBI ตั้งแต่เวอร์ชัน v 1.0.0 เป็นต้นไป การดาวน์โหลดข้อมูลจากแต่ละพื้นที่เก็บข้อมูลจะถูกแบ่งออกเป็นฟังก์ชันของตัวเอง นอกจากนี้ CRABS จะไม่จัดรูปแบบข้อมูลโดยอัตโนมัติหลังจากดาวน์โหลดเพื่อเพิ่มความยืดหยุ่นและเปิดใช้งานการดีบักเมื่อการดาวน์โหลดข้อมูลล้มเหลว
นอกจากการดาวน์โหลดข้อมูลลำดับแล้ว CRABS ยังสามารถดาวน์โหลดข้อมูลอนุกรมวิธาน NCBI ซึ่ง CRABS ใช้ในการสร้างลำดับวงศ์ตระกูลอนุกรมวิธานสำหรับแต่ละลำดับอีกด้วย
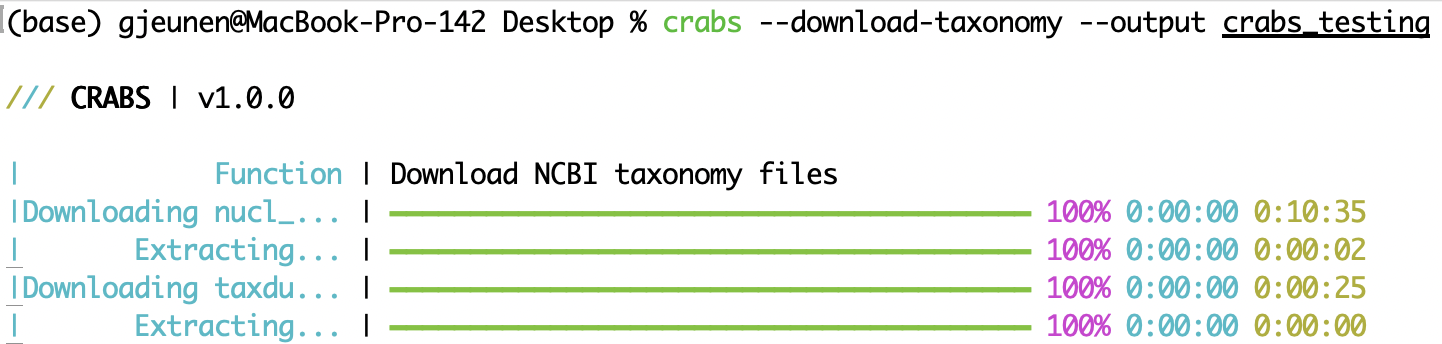
--download-taxonomy ในการกำหนดลำดับวงศ์ตระกูลอนุกรมวิธานให้กับแต่ละลำดับที่ดาวน์โหลดในฐานข้อมูลอ้างอิง (ดู 5.2 โมดูล 2) จำเป็นต้องดาวน์โหลดข้อมูลอนุกรมวิธาน CRABS ใช้อนุกรมวิธานของ NCBI และดาวน์โหลดไฟล์เฉพาะสามไฟล์ลงในคอมพิวเตอร์ของคุณ: (i) ไฟล์ที่เชื่อมโยงหมายเลขภาคยานุวัติกับรหัสอนุกรมวิธาน ( nucl_gb.accession2taxid ) (ii) ไฟล์ที่มีข้อมูลเกี่ยวกับชื่อสายวิวัฒนาการที่เกี่ยวข้องกับแต่ละรหัสอนุกรมวิธาน ( names.dmp ) และ (iii) ไฟล์ที่มีข้อมูลวิธีการเชื่อมโยงรหัสอนุกรมวิธาน ( nodes.dmp ) สามารถระบุไดเร็กทอรีเอาต์พุตสำหรับไฟล์ที่ดาวน์โหลดได้โดยใช้พารามิเตอร์ --output หากต้องการยกเว้นไฟล์ nucl_gb.accession2taxid หรือไฟล์ name.dmp และ nodes.dmp คุณสามารถระบุพารามิเตอร์ --exclude acc2tax หรือ --exclude taxdump ตามลำดับ รหัสแรกด้านล่างนี้ไม่ได้ดาวน์โหลดไฟล์ใดๆ เนื่องจากมีการระบุทั้ง acc2tax และ taxdump สำหรับพารามิเตอร์ --exclude โค้ดบรรทัดที่สองดาวน์โหลดไฟล์ทั้งสามไฟล์ไปยังไดเร็กทอรีย่อย --output crabs_testing ภาพหน้าจอด้านล่างแสดงสิ่งที่พิมพ์ไปยังคอนโซลเมื่อรันโค้ดบรรทัดนี้
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold ดาวน์โหลดลำดับ BOLD ผ่านทางเว็บไซต์ BOLD ไฟล์เอาต์พุตซึ่งมีโครงสร้างเป็นเอกสาร fasta สองบรรทัดสามารถระบุได้โดยใช้พารามิเตอร์ --output ผู้ใช้สามารถระบุกลุ่มอนุกรมวิธานที่จะดาวน์โหลดโดยใช้พารามิเตอร์ --taxon เราขอแนะนำให้เขียน for loop แบบง่าย (ตัวอย่างด้านล่าง) เมื่อผู้ใช้ต้องการดาวน์โหลดกลุ่มอนุกรมวิธานหลายกลุ่ม ดังนั้นจึงเป็นการจำกัดจำนวนข้อมูลจาก BOLD ต่ออินสแตนซ์ อย่างไรก็ตาม หากกลุ่มอนุกรมวิธานสนใจเพียงจำนวนจำกัด ชื่อกลุ่มอนุกรมวิธานก็สามารถคั่นด้วย | ได้เช่นกัน (ตัวอย่างด้านล่าง) นอกจากนี้เรายังแนะนำให้ผู้ใช้ตรวจสอบว่าชื่อกลุ่มอนุกรมวิธานที่จะดาวน์โหลดแสดงอยู่ในไฟล์เก็บถาวร BOLD หรือไม่ หรือจำเป็นต้องใช้ชื่ออื่นหรือไม่ ตัวอย่างเช่น การระบุ --taxon Chondrichthyes จะไม่ดาวน์โหลดลำดับปลากระดูกอ่อนทั้งหมดจาก BOLD เนื่องจากชื่อคลาสนี้ไม่อยู่ในรายการ BOLD ผู้ใช้ควรใช้ --taxon Elasmobranchii ในกรณีนี้ ผู้ใช้ยังสามารถระบุเพื่อจำกัดการดาวน์โหลดไว้ที่เครื่องหมายทางพันธุกรรมเฉพาะโดยระบุพารามิเตอร์ --marker เมื่อสนใจเครื่องหมายทางพันธุกรรมหลายตัว ชื่อเครื่องหมายควรคั่นด้วย | - เครื่องหมายบาร์โค้ด DNA หลักสี่ตัวบน BOLD ได้แก่ COI-5P , ITS , matK และ rbcL อินพุตสำหรับพารามิเตอร์ --marker คำนึงถึงขนาดตัวพิมพ์
วิธีการที่แนะนำ: for loop อย่างง่ายในการดาวน์โหลดข้อมูลจาก BOLD สำหรับกลุ่มอนุกรมวิธานหลายกลุ่ม (แนวทางที่แนะนำ) โค้ดด้านล่างจะดาวน์โหลดข้อมูลสำหรับ Elasmobranchii ก่อน ตามด้วยลำดับที่กำหนดให้กับ Mammalia ข้อมูลที่ดาวน์โหลดจะถูกเขียนลงในไดเร็กทอรีย่อย --output crabs_testing และวางไว้ในไฟล์สองไฟล์แยกกัน เพื่อระบุว่าข้อมูลใดอยู่ในกลุ่มอนุกรมวิธานใด เช่น crabs_testing/bold_Elasmobranchii.fasta และ crabs_testing/bold_Mammalia.fasta
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

ตัวเลือกอื่น: นอกจาก for loop ที่แนะนำแล้ว คุณสามารถระบุชื่อแท็กซอนได้หลายรายการพร้อมกันโดยแยกชื่อโดยใช้ | -
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl ลำดับจาก EMBL จะถูกดาวน์โหลดผ่านทางไซต์ ENA FTP ไฟล์ EMBL จะถูกดาวน์โหลดในรูปแบบ '.fasta.gz' ก่อน และจะถูกแตกไฟล์โดยอัตโนมัติเมื่อการดาวน์โหลดเสร็จสิ้น ฐานข้อมูลนี้ไม่มีความยืดหยุ่นในการดาวน์โหลดแบบเลือกมากนัก เมื่อเทียบกับ BOLD หรือ NCBI แต่ข้อมูล EMBL ถูกจัดโครงสร้างออกเป็น 15 แผนกภาษี ซึ่งสามารถดาวน์โหลดแยกกันได้ คุณสามารถระบุแผนกภาษีที่จะดาวน์โหลดได้โดยใช้พารามิเตอร์ --taxon เนื่องจากแต่ละแผนกภาษีแบ่งออกเป็นหลายไฟล์ จึงมี * ไว้หลังชื่อเพื่อดาวน์โหลดไฟล์ทั้งหมด ผู้ใช้ยังสามารถดาวน์โหลดไฟล์เฉพาะโดยการเขียนชื่อไฟล์แบบเต็ม รายการตัวเลือกแผนกภาษีทั้ง 15 ตัวเลือกมีดังต่อไปนี้ สามารถระบุไดเร็กทอรีเอาต์พุตและชื่อไฟล์ได้โดยใช้พารามิเตอร์ --output
รายชื่อแผนกภาษี:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS ยังสามารถดาวน์โหลดฐานข้อมูล MitoFish ได้ ฐานข้อมูลนี้เป็นไฟล์ fasta สองบรรทัดไฟล์เดียว สามารถระบุไดเร็กทอรีเอาต์พุตและชื่อไฟล์ได้โดยใช้พารามิเตอร์ --output
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi ลำดับจากฐานข้อมูล NCBI จะถูกดาวน์โหลดผ่าน Entrez Programming Utilities NCBI อนุญาตให้ดาวน์โหลดข้อมูลจากฐานข้อมูลต่างๆ ซึ่งผู้ใช้สามารถระบุด้วยพารามิเตอร์ --database สำหรับผู้ใช้ส่วนใหญ่ --database nucleotide จะเหมาะสมที่สุดสำหรับการสร้างฐานข้อมูลอ้างอิงในเครื่อง
ในการระบุข้อมูลที่จะดาวน์โหลดจาก NCBI ผู้ใช้จะทำการค้นหาผ่านพารามิเตอร์ --query การสร้างการค้นหา NCBI ที่ดีอาจเป็นเรื่องยาก วิธีที่ดีในการสร้างคำค้นหาคือการใช้หน้าต่างค้นหาหน้าเว็บของ NCBI จากลิงค์นี้ ให้ทำการค้นหาเบื้องต้นแล้วกด Enter สิ่งนี้จะนำคุณไปยังหน้าผลลัพธ์ซึ่งคุณสามารถปรับแต่งการค้นหาของคุณเพิ่มเติมได้ ในภาพหน้าจอด้านล่าง เราได้ปรับปรุงการค้นหาเพิ่มเติมโดยการจำกัดความยาวของลำดับระหว่าง 100 - 25,000 bp และรวมเฉพาะลำดับไมโตคอนเดรียเท่านั้น ผู้ใช้สามารถคัดลอกและวางข้อความในช่อง "รายละเอียดการค้นหา" บนเว็บไซต์ และใส่เครื่องหมายคำพูดให้กับพารามิเตอร์ --query ข้อดีอีกประการหนึ่งของการใช้หน้าต่างค้นหาหน้าเว็บของ NCBI ก็คือหน้าเว็บจะแสดงจำนวนลำดับที่ตรงกับคำค้นหาของคุณ ซึ่งควรตรงกับจำนวนลำดับที่รายงานโดย CRABS หน้าเว็บนี้มีบทช่วยสอนสั้นๆ เพิ่มเติมเกี่ยวกับการใช้ฟังก์ชันการค้นหาในหน้าเว็บ NCBI ที่ทีมงานของเราเขียนขึ้นเพื่อหาข้อมูลเพิ่มเติม
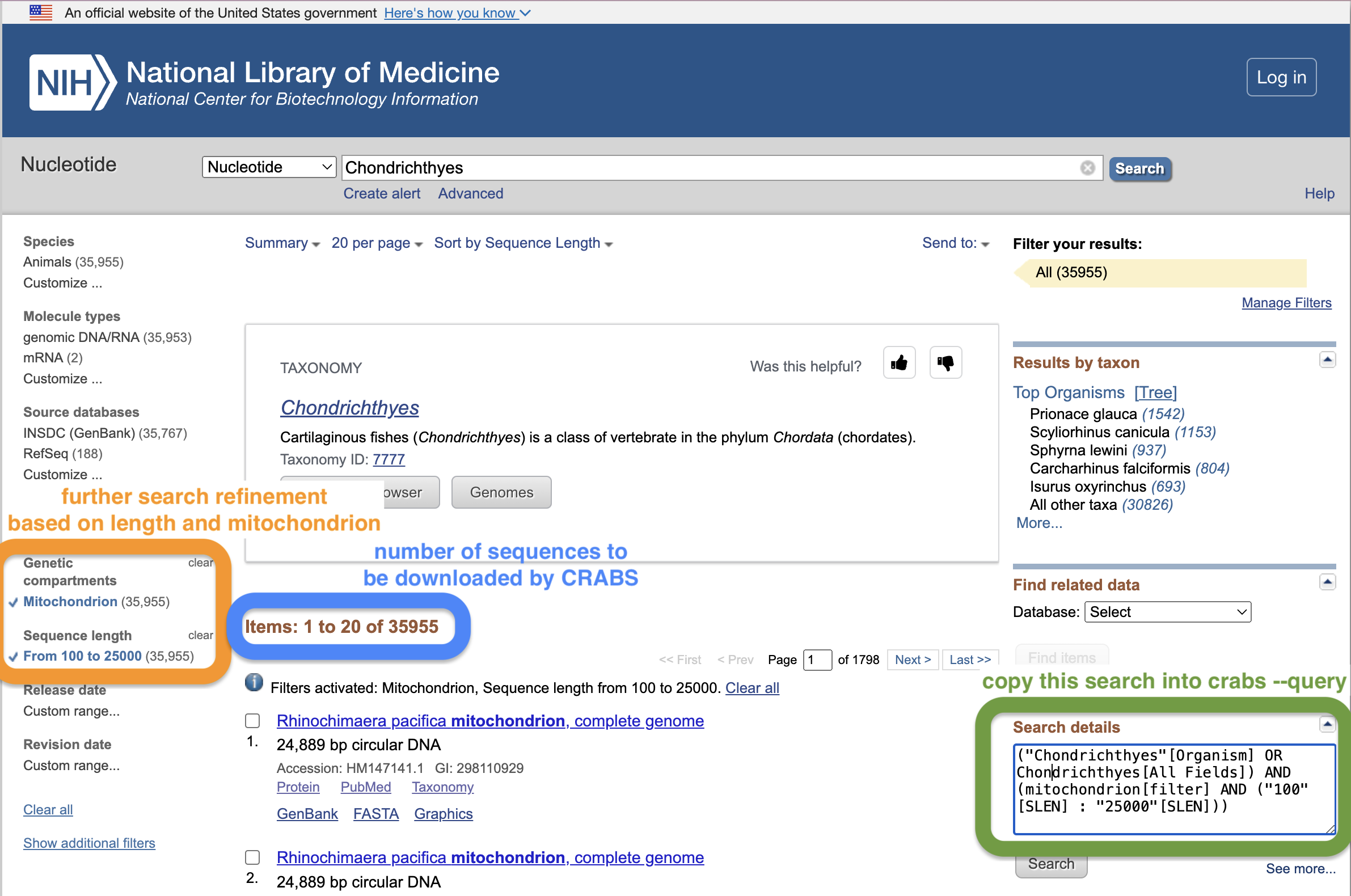
นอกจากคำค้นหา ( --query ) แล้ว ผู้ใช้ยังสามารถจำกัดคำค้นหาเพิ่มเติมได้โดยการดาวน์โหลดข้อมูลลำดับสำหรับรายการชนิดพันธุ์โดยใช้พารามิเตอร์ --species พารามิเตอร์ --species ใช้สตริงอินพุตของชื่อสายพันธุ์โดยคั่นด้วย + หรือไฟล์อินพุต .txt ที่มีชื่อสายพันธุ์เดียวต่อบรรทัดในเอกสาร พารามิเตอร์ --batchsize ให้ตัวเลือกแก่ผู้ใช้ในการดาวน์โหลดลำดับเป็นชุดของ N จากเว็บไซต์ NCBI พารามิเตอร์นี้มีค่าเริ่มต้นอยู่ที่ 5,000 ไม่แนะนำให้เพิ่มค่านี้เกินกว่า 5,000 เนื่องจากเซิร์ฟเวอร์ NCBI มักจะยกเลิกการดาวน์โหลดหากมีการดาวน์โหลดลำดับมากเกินไปในครั้งเดียว พารามิเตอร์ --email อนุญาตให้ผู้ใช้ระบุที่อยู่อีเมลของตน ซึ่งจำเป็นในการเข้าถึงเซิร์ฟเวอร์ NCBI ในที่สุด ไดเร็กทอรีเอาต์พุตและชื่อไฟล์สามารถระบุได้โดยใช้พารามิเตอร์ --output
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import เมื่อดาวน์โหลดไฟล์จากที่เก็บข้อมูลออนไลน์แล้ว ไฟล์จะต้องนำเข้าไปยัง CRABS โดยใช้ฟังก์ชัน --import รูปแบบ CRABS ประกอบด้วยบรรทัดที่คั่นด้วยแท็บเดียวตามลำดับที่มีข้อมูลทั้งหมด รวมถึง (i) รหัสลำดับ (ii) ชื่ออนุกรมวิธานที่แยกวิเคราะห์จากการดาวน์โหลดครั้งแรก (iii) หมายเลขรหัสอนุกรมวิธาน NCBI (iv) เชื้อสายอนุกรมวิธานตามอนุกรมวิธานของ NCBI และ (v) ลำดับ CRABS จะพยายามรับหมายเลขภาคยานุวัติ NCBI สำหรับแต่ละลำดับเป็นรหัสลำดับ หากลำดับไม่มีหมายเลขภาคยานุวัติ กล่าวคือ ไม่ได้ฝากไว้ใน NCBI CRABS จะสร้าง ID ลำดับที่ไม่ซ้ำกันโดยใช้รูปแบบต่อไปนี้: crabs_*[num]*_taxonomic_name รูปแบบของเอกสารอินพุตถูกระบุโดยใช้พารามิเตอร์ --import-format และระบุชื่อของพื้นที่เก็บข้อมูลที่ใช้ดาวน์โหลดข้อมูล เช่น BOLD , EMBL , MITOFISH หรือ NCBI CRABS ลำดับวงศ์ตระกูลอนุกรมวิธานที่สร้างขึ้นนั้นอิงตามอนุกรมวิธานของ NCBI และ CRABS ต้องการไฟล์สามไฟล์ที่ดาวน์โหลดโดยใช้ฟังก์ชัน --download-taxonomy กล่าวคือ --names , --nodes และ --acc2tax จากเวอร์ชัน v 1.0.0 CRABS สามารถแก้ไขคำพ้องความหมายและชื่อที่ไม่ได้รับการยอมรับ เพื่อรวมลำดับและความหลากหลายจำนวนมากขึ้นในฐานข้อมูลอ้างอิงในท้องถิ่น อันดับอนุกรมวิธานที่จะรวมไว้ในเชื้อสายอนุกรมวิธานสามารถระบุได้โดยใช้พารามิเตอร์ --ranks แม้ว่าจะสามารถรวมอันดับอนุกรมวิธานใดๆ ก็ตามได้ แต่เราขอแนะนำให้ใช้ข้อมูลต่อไปนี้เพื่อรวมข้อมูลที่จำเป็นทั้งหมดสำหรับตัวแยกประเภทอนุกรมวิธานส่วนใหญ่ --ranks 'superkingdom;phylum;class;order;family;genus;species' ไฟล์เอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --output และเป็นไฟล์ .txt แบบธรรมดา ในหน้าต่างเทอร์มินัล CRABS จะพิมพ์ผลลัพธ์ของจำนวนลำดับที่นำเข้า รวมถึงลำดับใดๆ ที่ไม่สามารถสร้างลำดับอนุกรมวิธานได้
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

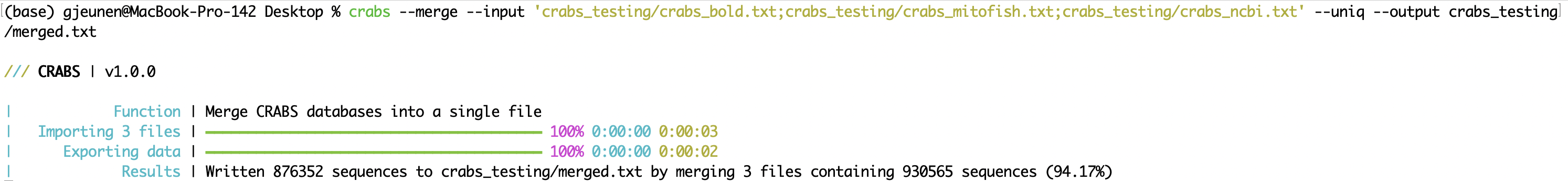
--merge เมื่อดาวน์โหลดข้อมูลลำดับจากที่เก็บข้อมูลออนไลน์หลายแห่ง ไฟล์สามารถรวมเป็นไฟล์เดียวได้หลังจากการนำเข้า (ดู 5.2.1 --import ) โดยใช้ฟังก์ชัน --merge ไฟล์อินพุตที่จะรวมสามารถป้อนได้โดยใช้พารามิเตอร์ --input โดยคั่นไฟล์ด้วย ; - เป็นไปได้ว่ามีการดาวน์โหลดลำดับหลายครั้งเมื่อฝากไว้ในที่เก็บข้อมูลออนไลน์ต่างๆ การใช้พารามิเตอร์ --uniq จะรักษาหมายเลขภาคยานุวัติแต่ละหมายเลขไว้เพียงเวอร์ชันเดียว ไฟล์เอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --output ในหน้าต่างเทอร์มินัล CRABS จะพิมพ์ผลลัพธ์ของจำนวนลำดับที่ผสาน เช่นเดียวกับจำนวนลำดับที่คงไว้เมื่อใช้พารามิเตอร์ --uniq
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
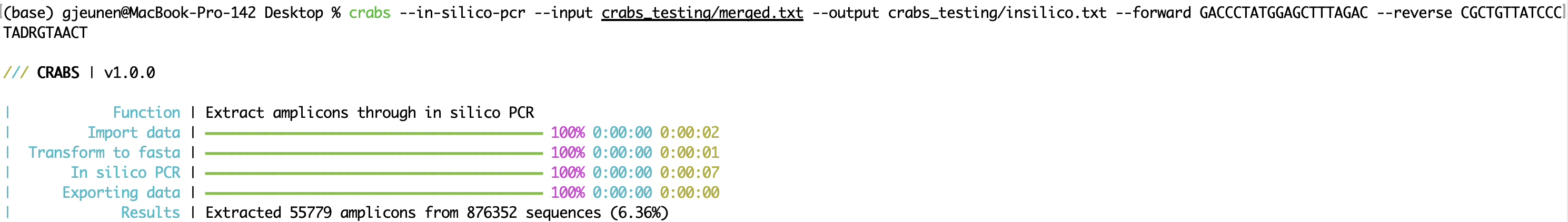
CRABS แยกบริเวณแอมพลิคอนของไพรเมอร์ที่ตั้งค่าไว้โดยการดำเนินการ in silico PCR (ฟังก์ชัน: --in-silico-pcr ) CRABS ใช้ cutadapt v 4.4 สำหรับ in silico PCR เพื่อเพิ่มความเร็วในการประมวลผลโค้ด Python แบบเดิม ชื่อไฟล์อินพุตและเอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ ' --input ' และ ' --output ' ตามลำดับ ควรจัดเตรียมไพรเมอร์ทั้งแบบเดินหน้าและถอยหลังในทิศทาง 5'-3' โดยใช้พารามิเตอร์ ' --forward ' และ ' --reverse ' ตามลำดับ CRABS จะทำการเสริมไพรเมอร์แบบย้อนกลับ ตั้งแต่เวอร์ชัน v 1.0.0 CRABS สามารถรักษาบาร์โค้ดได้ทั้งสองทิศทางโดยใช้การวิเคราะห์ PCR แบบซิลิโก เพียงตัวเดียว ดังนั้นจึงไม่มีขั้นตอนการเสริมแบบย้อนกลับและการรันซ้ำของ in silico PCR ดังนั้นจึงเพิ่มความเร็วในการดำเนินการได้อย่างมาก หากต้องการรักษาลำดับซึ่งไม่พบขอบเขตที่มีผลผูกพันไพรเมอร์ สามารถระบุไฟล์เอาต์พุตสำหรับพารามิเตอร์ --untrimmed จำนวนที่ไม่ตรงกันสูงสุดที่อนุญาตซึ่งพบในบริเวณที่มีผลผูกพันกับไพรเมอร์สามารถระบุได้โดยใช้พารามิเตอร์ --mismatch โดยมีการตั้งค่าเริ่มต้นที่ 4 สุดท้ายนี้ การวิเคราะห์ in silico PCR สามารถเป็นแบบมัลติเธรดใน CRABS ตามค่าเริ่มต้น มีการใช้จำนวนเธรดสูงสุด แต่ผู้ใช้สามารถระบุจำนวนเธรดที่จะใช้กับพารามิเตอร์ --threads
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
เป็นเรื่องปกติที่จะลบขอบเขตที่มีผลผูกพันกับไพรเมอร์ออกจากลำดับการอ้างอิงเมื่อฝากไว้ในฐานข้อมูลออนไลน์ ดังนั้น เมื่อลำดับการอ้างอิงถูกสร้างขึ้นโดยใช้ไพรเมอร์ไปข้างหน้าและ/หรือย้อนกลับเดียวกันกับที่ค้นหาในฟังก์ชัน --in-silico-pcr ฟังก์ชัน --in-silico-pcr จะล้มเหลวในการกู้คืนขอบเขตแอมพลิคอนของ ลำดับการอ้างอิง เพื่อพิจารณาถึงความเป็นไปได้นี้ CRABS มีตัวเลือกในการเรียกใช้การจัดตำแหน่งแบบคู่ตามลำดับทั่วโลก ซึ่งใช้งานโดยใช้ VSEARCH v 2.16.0 เพื่อแยกขอบเขตแอมพลิคอนซึ่งลำดับการอ้างอิงไม่มีขอบเขตที่มีผลผูกพันไพรเมอร์แบบเต็มไปข้างหน้าและย้อนกลับ เพื่อให้บรรลุเป้าหมายนี้ ฟังก์ชัน --pairwise-global-alignment จะใช้ไฟล์ฐานข้อมูลที่ดาวน์โหลดมาแต่แรกโดยใช้พารามิเตอร์ --input ฐานข้อมูลที่จะค้นหาคือไฟล์เอาต์พุตจาก --in-silico-pcr และสามารถระบุได้โดยใช้พารามิเตอร์ --amplicons ไฟล์เอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --output ลำดับไพรเมอร์ซึ่งใช้ในการคำนวณความยาวคู่เบสเท่านั้น สามารถตั้งค่าได้ด้วยพารามิเตอร์ --forward และ --reverse เนื่องจากฟังก์ชัน --pairwise-global-alignment อาจใช้เวลานานในการรันสำหรับฐานข้อมูลขนาดใหญ่ ความยาวลำดับจึงสามารถจำกัดได้เพื่อเร่งกระบวนการโดยใช้พารามิเตอร์ --size-select เปอร์เซ็นต์ขั้นต่ำของการระบุตัวตนและความครอบคลุมของการค้นหาสามารถระบุได้โดยใช้พารามิเตอร์ --percent-identity และ --coverage ตามลำดับ --percent-identity ควรระบุเป็นค่าเปอร์เซ็นต์ระหว่าง 0 ถึง 1 (เช่น 95% = 0.95) ในขณะที่ --coverage ควรระบุเป็นค่าเปอร์เซ็นต์ระหว่าง 0 ถึง 100 (เช่น 95% = 95) ตามค่าเริ่มต้น ฟังก์ชัน --pairwise-global-alignment ถูกจำกัดไว้เพื่อรักษาลำดับโดยที่ลำดับไพรเมอร์ไม่ปรากฏครบถ้วนในลำดับอ้างอิง (การจัดตำแหน่งเริ่มต้นหรือสิ้นสุดภายในความยาวของไพรเมอร์ไปข้างหน้าหรือย้อนกลับ) เมื่อระบุพารามิเตอร์ --all-start-positions การเข้าชมที่เป็นบวกจะถูกรวมไว้เมื่อพบการจัดตำแหน่งนอกช่วงของขอบเขตที่มีผลผูกพันกับไพรเมอร์ (พลาดโดยฟังก์ชัน --in-silico-pcr เนื่องจากมีความไม่ตรงกันมากเกินไปใน บริเวณที่มีผลผูกพันกับไพรเมอร์) เราไม่แนะนำให้ใช้ --all-start-positions เนื่องจากมีโอกาสน้อยมากที่บาร์โค้ดจะถูกขยายโดยใช้ชุดไพรเมอร์ที่ระบุของฟังก์ชัน --in-silico-pcr เมื่อมีข้อมูลที่ไม่ตรงกันมากกว่า 4 รายการในไพรเมอร์- ภูมิภาคที่มีผลผูกพัน
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
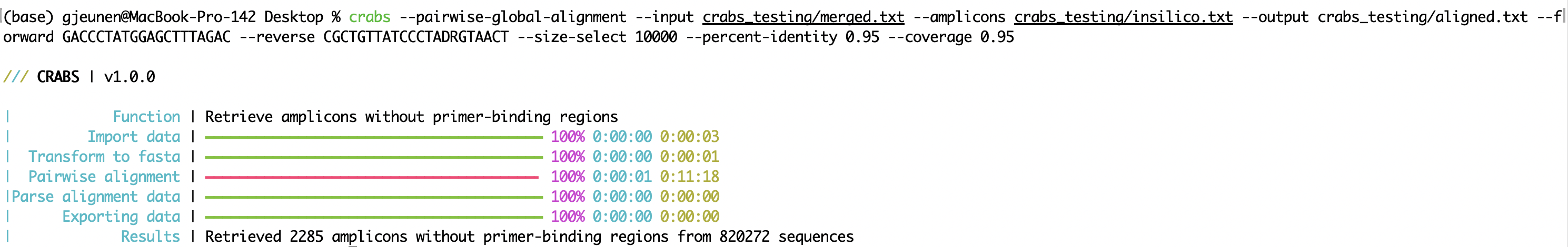
--pairwise-global-alignment ฟังก์ชัน --pairwise-global-alignment อาจใช้เวลานานในการดำเนินการเมื่อ CRABS กำลังประมวลผลไฟล์ลำดับขนาดใหญ่ แม้ว่าจะรองรับมัลติเธรดก็ตาม นับตั้งแต่อัปเดตเป็น CRABS v 1.0.0 โครงสร้างไฟล์ที่เหมือนกันก็ถูกนำมาใช้ตั้งแต่ --import ถึง --export ซึ่งช่วยให้สามารถดำเนินการฟังก์ชันต่างๆ ในลำดับใดก็ได้ แม้ว่าเรายังคงแนะนำให้ปฏิบัติตามลำดับของเวิร์กโฟลว์ CRABS แต่ฟังก์ชัน --pairwise-global-alignment สามารถเร่งความเร็วได้อย่างมากเมื่อดำเนินการฟังก์ชัน --dereplicate และ --filter ก่อนฟังก์ชัน --in-silico-pcr ด้วยการดำเนินขั้นตอนการดูแลจัดการเหล่านี้ก่อน --in-silico-pcr จำนวนลำดับที่จำเป็นในการประมวลผลโดย CRABS สำหรับฟังก์ชัน --pairwise-global-alignment จะลดลงอย่างมาก
หมายเหตุ 1 : เมื่อดำเนินการฟังก์ชัน --filter ก่อน --in-silico-pcr โปรดตรวจสอบให้แน่ใจว่าได้ละเว้นพารามิเตอร์ใดๆ ที่ส่งผลกระทบโดยตรงต่อลำดับ เนื่องจาก --filter จะใช้พื้นฐานนี้กับลำดับทั้งหมด ไม่ใช่แอมพลิคอนที่แยกออกมา . ดังนั้น ละเว้นพารามิเตอร์ต่อไปนี้: --minimum-length , --maximum-length , --maximum-n
หมายเหตุ 2 : เมื่อดำเนินการฟังก์ชัน --dereplicate และ --filter ก่อน --in-silico-pcr ขอแนะนำให้เรียกใช้ฟังก์ชันทั้งสองอีกครั้งหลังจาก --pairwise-global-alignment เนื่องจากฐานข้อมูลสามารถดูแลจัดการเพิ่มเติมได้ในขณะนี้ ว่าแอมพลิคอนจะถูกแยกออกมา
เมื่อบาร์โค้ดที่เป็นไปได้ทั้งหมดสำหรับชุดไพรเมอร์ได้รับการแยกออกโดยฟังก์ชัน --in-silico-pcr และ --pairwise-global-alignment ฐานข้อมูลอ้างอิงเฉพาะที่สามารถรับการดูแลจัดการเพิ่มเติมและการตั้งค่าย่อยภายใน CRABS โดยใช้ฟังก์ชันต่างๆ รวมถึง --dereplicate , --filter และ --subset
--dereplicate วิธีแรกในการดูแลจัดการคือการจำลองฐานข้อมูลอ้างอิงในเครื่องโดยใช้ฟังก์ชัน --dereplicate อาจเป็นไปได้ว่า ณ จุดนี้บาร์โค้ดที่เหมือนกันหลายแท็กซ่าจะอยู่ในฐานข้อมูลอ้างอิงในพื้นที่ สิ่งนี้สามารถเกิดขึ้นได้เมื่อกลุ่มการวิจัยต่างๆ ได้ฝากลำดับที่เหมือนกันไว้ หรือหากความแปรผันภายในเฉพาะระหว่างลำดับสำหรับอนุกรมวิธานไม่มีอยู่ในบาร์โค้ดที่แยกออกมา เป็นการดีที่สุดที่จะลบบาร์โค้ดอ้างอิงที่เหมือนกันเหล่านี้ออกเพื่อเร่งการกำหนดอนุกรมวิธาน ตลอดจนปรับปรุงผลลัพธ์การกำหนดอนุกรมวิธาน (โดยเฉพาะอย่างยิ่งสำหรับตัวแยกประเภทอนุกรมวิธานที่ให้ผลลัพธ์ในจำนวนจำกัด เช่น BLAST)
ไฟล์อินพุตและเอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --input และ --output ตามลำดับ CRABS เสนอวิธีการจำลองแบบสามวิธี ซึ่งสามารถระบุได้โดยใช้พารามิเตอร์ --dereplication-method ได้แก่:
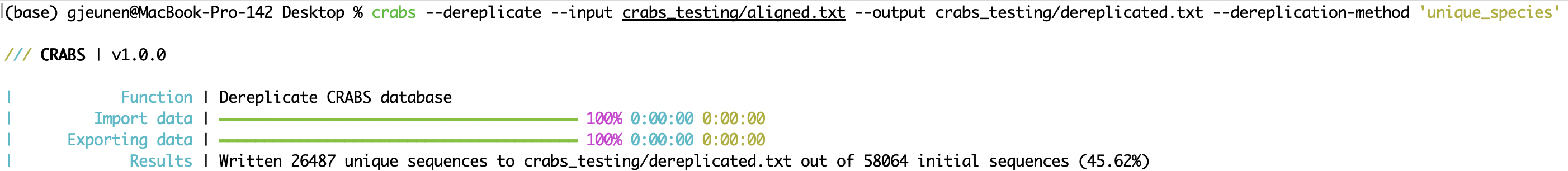
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
--filter วิธีการดูแลที่สองคือการกรองฐานข้อมูลอ้างอิงในเครื่องโดยใช้พารามิเตอร์ต่างๆ โดยใช้ฟังก์ชัน --filter ไฟล์อินพุตและเอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --input และ --output ตามลำดับ จากเวอร์ชัน v 1.0.0 CRABS รวมการกรองตามพารามิเตอร์หกตัว ได้แก่:
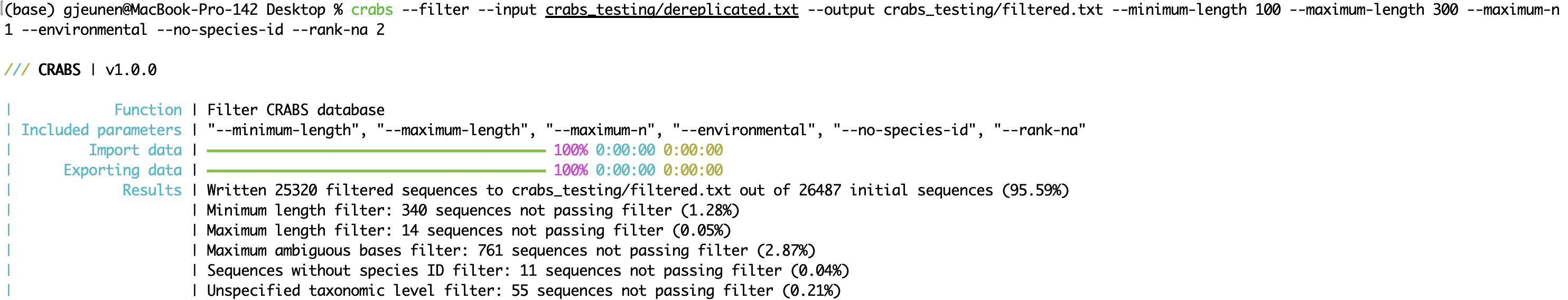
--minimum-length : ความยาวลำดับขั้นต่ำสำหรับเครื่องขยายเสียงที่จะเก็บไว้ในฐานข้อมูล--maximum-length : ความยาวลำดับสูงสุดสำหรับเครื่องขยายเสียงที่จะเก็บไว้ในฐานข้อมูล--maximum-n : ละทิ้งแอมพลิคอนที่มีฐาน N หรือมากกว่าที่คลุมเครือ ( N );--environmental : ละทิ้งลำดับสิ่งแวดล้อมจากฐานข้อมูล--no-species-id : ละทิ้งลำดับที่ไม่มีชื่อสายพันธุ์--rank-na : ละทิ้งลำดับที่มีระดับอนุกรมวิธานที่ไม่ระบุตั้งแต่ N ขึ้นไป crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset วิธีการดูแลจัดการที่สามและสุดท้ายที่รวมอยู่ใน CRABS คือการจัดย่อยฐานข้อมูลอ้างอิงในพื้นที่เพื่อรวม (พารามิเตอร์: --include ) หรือไม่รวม (พารามิเตอร์: --exclude ) แท็กซ่าเฉพาะโดยใช้ฟังก์ชัน --subset ฟังก์ชันนี้ช่วยให้สามารถลบบาร์โค้ดอ้างอิงออกจากกลุ่มอนุกรมวิธานที่ไม่เกี่ยวข้องกับคำถามการวิจัยได้ กลุ่มอนุกรมวิธานเหล่านี้อาจถูกรวมไว้ในฐานข้อมูลอ้างอิงในพื้นที่เนื่องจากอาจมีการขยายชุดไพรเมอร์ที่ไม่เฉพาะเจาะจง กรณีการใช้งานอื่นสำหรับ --subset คือการลบลำดับที่ผิดพลาดที่ทราบ
สำหรับตัวแยกประเภทอนุกรมวิธานตามการเรียนรู้ของเครื่องจักร (IDTAXA) หรือระยะทาง k-mer (SINTAX) อาจเป็นประโยชน์ในการสับเซ็ตฐานข้อมูลอ้างอิงโดยรวมแท็กซ่าที่ทราบว่าเกิดขึ้นในภูมิภาคที่มีการเก็บตัวอย่าง และไม่รวมชนิดพันธุ์ที่เกี่ยวข้องอย่างใกล้ชิดซึ่งไม่ทราบว่าไม่ ที่จะเกิดขึ้นในภูมิภาคเพื่อเพิ่มความละเอียดทางอนุกรมวิธานที่ได้รับของตัวแยกประเภทเหล่านี้ และได้รับผลลัพธ์การกำหนดอนุกรมวิธานที่ดีขึ้น
ไฟล์อินพุตและเอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --input และ --output ตามลำดับ พารามิเตอร์ --include และ --exclude สามารถใช้ในรายการแท็กซ่าที่คั่นด้วย ; หรือไฟล์ .txt ที่มีชื่อแท็กซอนรายการเดียวต่อบรรทัด
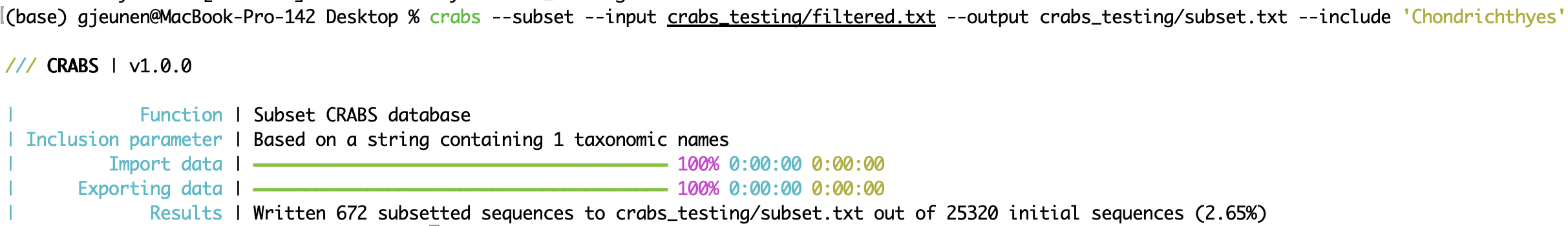
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
เมื่อฐานข้อมูลอ้างอิงเสร็จสิ้นแล้ว ก็สามารถส่งออกเป็นรูปแบบต่างๆ เพื่อรองรับข้อกำหนดที่กำหนดโดยเครื่องมือซอฟต์แวร์ส่วนใหญ่ในการกำหนดอนุกรมวิธานให้กับข้อมูลเมทาโนมิก ไฟล์อินพุตและเอาต์พุตสามารถระบุได้โดยใช้พารามิเตอร์ --input และ --output ตามลำดับ จากเวอร์ชัน v 1.0.0 CRABS ได้รวมการจัดรูปแบบของฐานข้อมูลอ้างอิงสำหรับตัวแยกประเภทที่แตกต่างกันหกตัว (พารามิเตอร์: --export-format ) ได้แก่:
--export-format 'sintax' : ตัวแยกประเภท SINTAX รวมอยู่ใน USEARCH และ VSEARCH;--export-format 'rdp' : ตัวแยกประเภท RDP เป็นโปรแกรมแบบสแตนด์อโลนที่ใช้กันอย่างแพร่หลายในการศึกษาไมโครไบโอม--export-format 'qiime-fasta' และ --export-format 'qiime-text' : สามารถใช้เพื่อกำหนดรหัสอนุกรมวิธานใน QIIME และ QIIME2;--export-format 'dada2-species' และ --export-format 'dada2-taxonomy' : สามารถใช้เพื่อกำหนดรหัสอนุกรมวิธานใน DADA2;--export-format 'idt-fasta' และ --export-format 'idt-text' : ตัวแยกประเภท IDTAXA เป็นอัลกอริธึมการเรียนรู้ของเครื่องที่รวมอยู่ในแพ็คเกจ DECIPHER R--export-format 'blast-notax' : สร้างฐานข้อมูลอ้างอิง BLAST ในท้องถิ่นสำหรับ Blastn และ Megablast ที่เอาต์พุตไม่ได้ให้รหัสอนุกรมวิธาน แต่แสดงรายการหมายเลขภาคยานุวัติ--export-format 'blast-tax' : สร้างฐานข้อมูลอ้างอิง BLAST ในท้องถิ่นสำหรับ Blastn และ Megablast ซึ่งเอาต์พุตให้ทั้งรหัสอนุกรมวิธานและหมายเลขภาคยานุวัติ crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

ในขณะที่การส่งออกฐานข้อมูลอ้างอิงท้องถิ่นไปยังรูปแบบเดียว (ยกเว้นตัวแยกประเภทที่ฐานข้อมูลอ้างอิงถูกแบ่งออกเป็นหลายไฟล์เช่น Qiime, Dada2, Idtaxa) จะเพียงพอสำหรับผู้ใช้ส่วนใหญ่ ฐานข้อมูลอ้างอิงไปยังหลายรูปแบบหากผู้ใช้ต้องการเปรียบเทียบผลลัพธ์ระหว่างตัวแยกประเภทอนุกรมวิธานที่แตกต่างกัน ตัวอย่างมีให้ด้านล่างเพื่อส่งออกฐานข้อมูลอ้างอิงท้องถิ่นในรูปแบบ Sintax, RDP และ IDTaxa
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

เมื่อฐานข้อมูลอ้างอิงเสร็จสิ้นแล้วปูสามารถเรียกใช้ฟังก์ชั่นหลังการประมวลผลห้าฟังก์ชั่นเพื่อสำรวจและให้ภาพรวมสรุปของฐานข้อมูลอ้างอิงในท้องถิ่นรวมถึง (i) --diversity-figure --amplicon-length-figure (ii) iii) --phylogenetic-tree , (iv) --amplification-efficiency-figure และ (v) --completeness-table
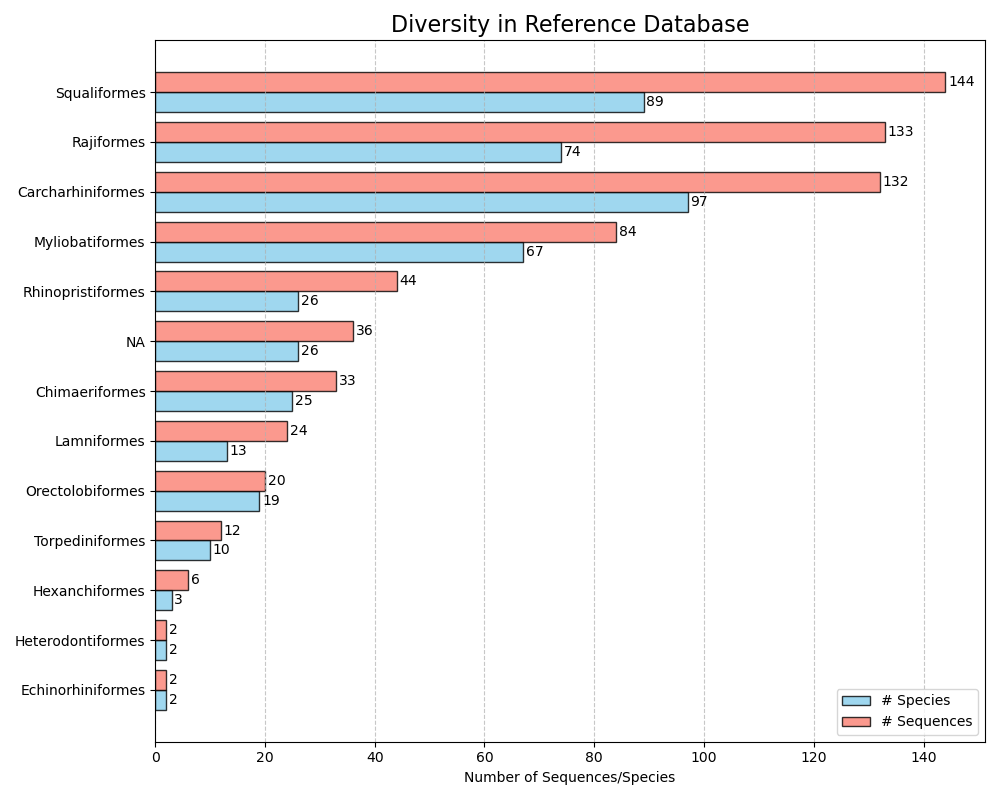
--diversity-figure ฟังก์ --diversity-figure แบบสร้างพล็อตบาร์แนวนอนที่มีจำนวนสปีชีส์ (สีน้ำเงิน) และจำนวนลำดับ (เป็นสีส้ม) ต่อสำหรับแต่ละกลุ่มอนุกรมวิธานในฐานข้อมูลอ้างอิง ผู้ใช้สามารถระบุอันดับอนุกรมวิธานเพื่อแยกฐานข้อมูลอ้างอิงด้วยพารามิเตอร์ --tax-level ระดับภาษีคือจำนวนของอันดับที่ปรากฏในระหว่างฟังก์ชัน --import ตัวอย่างเช่นถ้า --ranks 'superkingdom;phylum;class;order;family;genus;species' ถูกใช้ในระหว่าง --import การแยกออกจาก Superkingdom จะต้องใช้ --tax-level 1 , phylum = --tax-level 2 , class = --tax-level 3 ฯลฯ ไฟล์อินพุตในรูปแบบปูสามารถระบุได้โดยใช้พารามิเตอร์ --input รูปในรูปแบบ. png จะถูกเขียนไปยังไฟล์เอาต์พุตซึ่งสามารถระบุได้โดยใช้พารามิเตอร์ --output
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
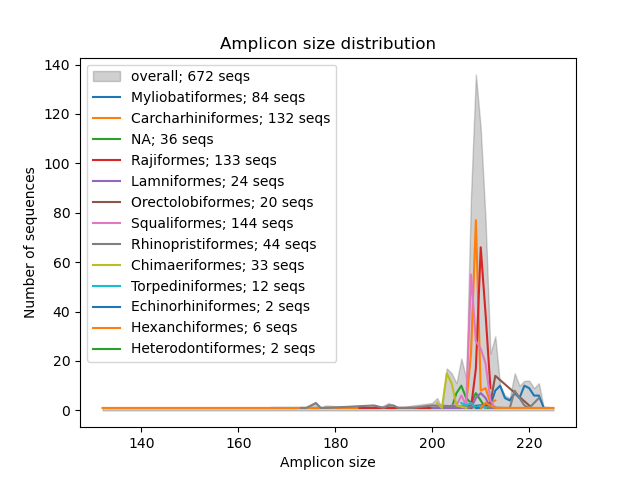
--amplicon-length-figure ฟังก์ชั่น --amplicon-length-figure ป์สร้างกราฟบรรทัดที่แสดงช่วงของความยาวแอมพลิฟายเออร์ ช่วงโดยรวมของความยาวแอมพลิฟายเออร์ในทุกลำดับในฐานข้อมูลอ้างอิงจะแสดงเป็นสีเทาแรเงาในขณะที่ผลลัพธ์แยกตามกลุ่มอนุกรมวิธาน (พารามิเตอร์: --tax-level ) ถูกซ้อนทับด้วยเส้นสี นอกจากนี้ตำนานจะแสดงจำนวนลำดับที่กำหนดให้กับแต่ละกลุ่มอนุกรมวิธานและจำนวนทั้งหมดของลำดับในฐานข้อมูลอ้างอิง ไฟล์อินพุตในรูปแบบปูสามารถระบุได้โดยใช้พารามิเตอร์ --input รูปในรูปแบบ. png จะถูกเขียนไปยังไฟล์เอาต์พุตซึ่งสามารถระบุได้โดยใช้พารามิเตอร์ --output
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
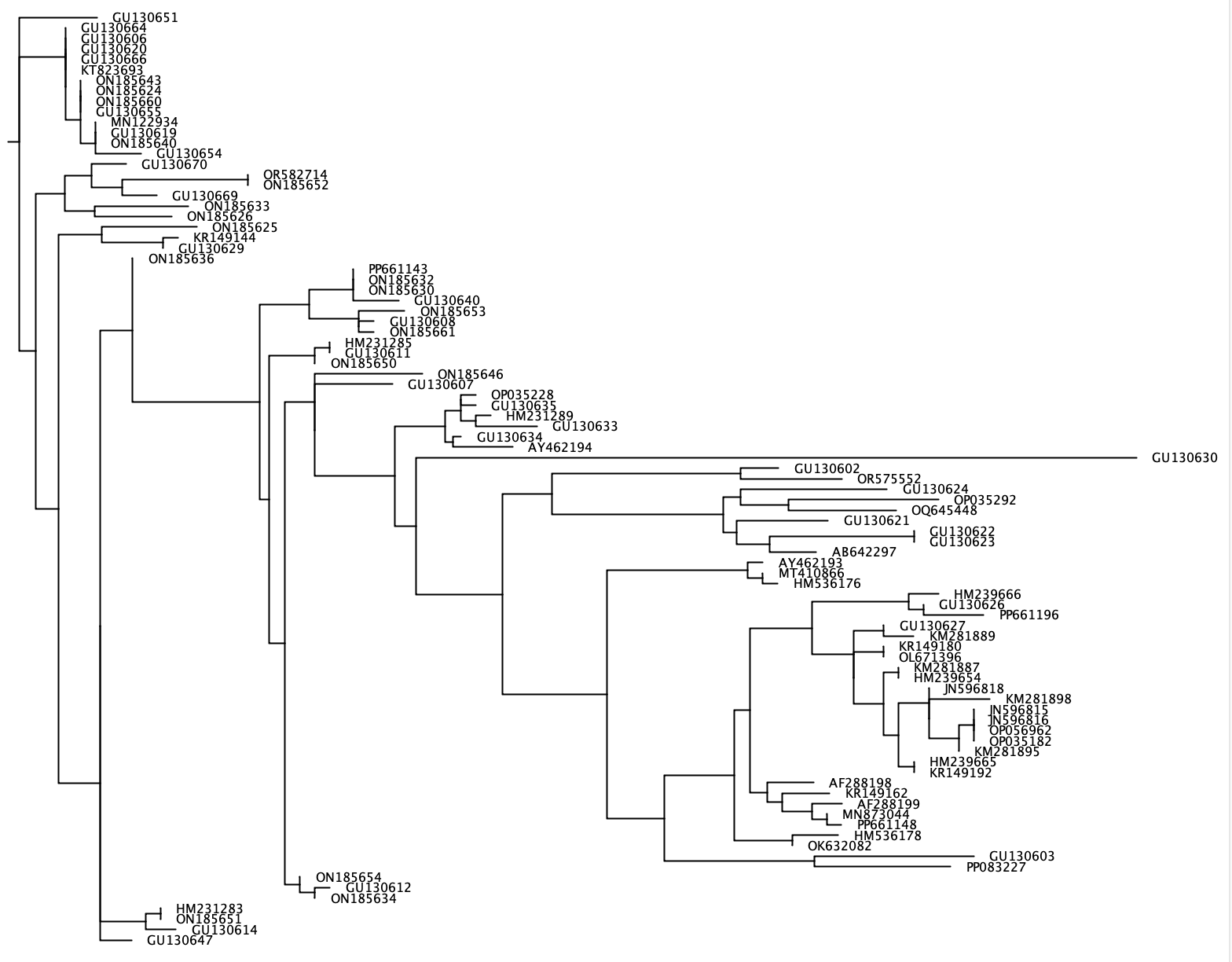
--phylogenetic-tree ฟังก์ชัน --phylogenetic-tree จะสร้างต้นไม้สายวิวัฒนาการสำหรับรายการสายพันธุ์ที่น่าสนใจ รายการของสายพันธุ์ที่น่าสนใจนี้สามารถนำเข้าได้โดยใช้พารามิเตอร์ --species และประกอบด้วยสตริงอินพุตคั่นด้วย + หรือไฟล์. txt ที่มีชื่อสปีชีส์เดียวในแต่ละบรรทัด สำหรับแต่ละสปีชีส์ที่น่าสนใจลำดับจะถูกสกัดจากฐานข้อมูลอ้างอิงที่แบ่งปันอันดับอนุกรมวิธานที่ผู้ใช้กำหนด (พารามิเตอร์: --tax-level ) กับสายพันธุ์ที่น่าสนใจ ปูจะสร้างการจัดตำแหน่งของลำดับที่สกัดทั้งหมดโดยใช้ ClustalW2 V 2.1 และสร้างต้นไม้สายวิวัฒนาการที่เข้าร่วมโดยใช้ FastTree แผนผังสายวิวัฒนาการในรูปแบบ newick จะถูกเขียนไปยังไฟล์เอาต์พุตโดยใช้พารามิเตอร์ --output และสามารถมองเห็นได้ในโปรแกรมซอฟต์แวร์เช่น Figtree หรือ Geneious เนื่องจากต้นไม้สายวิวัฒนาการแยกต่างหากจะถูกสร้างขึ้นสำหรับแต่ละสปีชีส์ที่น่าสนใจ --output ใช้ในชื่อไฟล์ทั่วไปในขณะที่ไฟล์เอาต์พุตที่แน่นอนจะมีชื่อสามัญนี้ตามด้วย '_species_name.tree'
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
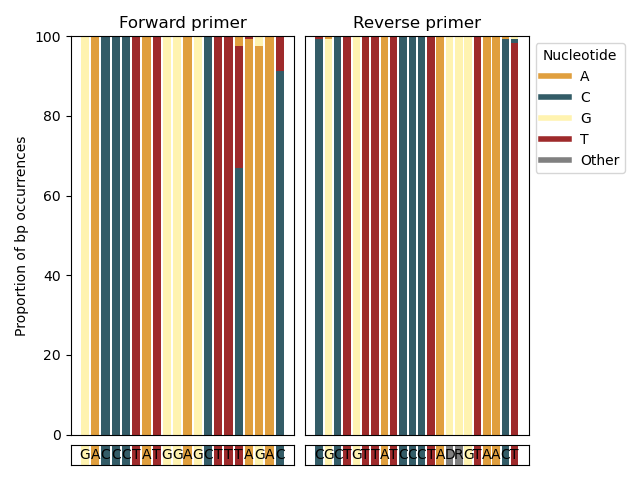
--amplification-efficiency-figure ฟังก์ --amplification-efficiency-figure จะสร้างกราฟแท่งโดยแสดงสัดส่วนของการเกิดขึ้นคู่ฐานในภูมิภาคที่มีผลผูกพันไพรเมอร์สำหรับกลุ่มอนุกรมวิธานที่ผู้ใช้ระบุ อาจเกิดขึ้นในกลุ่มที่น่าสนใจอนุกรมวิธานซึ่งอาจมีอิทธิพลต่อประสิทธิภาพการขยาย ฟังก์ชั่น --amplification-efficiency-figure ใช้ฐานข้อมูลอ้างอิงแบบปูขั้นสุดท้ายเป็นอินพุตโดยใช้พารามิเตอร์ --amplicons ในการค้นหาข้อมูลในภูมิภาคที่มีผลผูกพันไพรเมอร์สำหรับแต่ละลำดับในไฟล์อินพุตลำดับที่ดาวน์โหลดมาเริ่มต้นหลังจากจำเป็นต้องมีการนำเข้าโดยใช้พารามิเตอร์ --input ลำดับไพรเมอร์ไปข้างหน้าและย้อนกลับ (ในทิศทาง 5 ' -3') มีให้โดยใช้พารามิเตอร์ --forward และ --reverse ชื่อของกลุ่มที่น่าสนใจอนุกรมวิธานสามารถให้ได้โดยใช้พารามิเตอร์ --tax-group และสามารถตั้งค่าในระดับอนุกรมวิธานใด ๆ ที่รวมอยู่ในไฟล์อินพุต ในที่สุดรูปในรูปแบบ. PNG จะถูกเขียนไปยังไฟล์เอาต์พุตที่ระบุโดยพารามิเตอร์ --output
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table ฟังก์ --completeness-table จะส่งออกตารางที่คั่นด้วยแท็บ (พารามิเตอร์: --output ) พร้อมข้อมูลเกี่ยวกับรายการสายพันธุ์ที่น่าสนใจ รายการของสายพันธุ์ที่น่าสนใจนี้สามารถนำเข้าได้โดยใช้พารามิเตอร์ --species และประกอบด้วยสตริงอินพุตคั่นด้วย + หรือไฟล์. txt ที่มีชื่อสปีชีส์เดียวในแต่ละบรรทัด เชื้อสายอนุกรมวิธานจะถูกสร้างขึ้นสำหรับแต่ละสายพันธุ์ที่น่าสนใจโดยใช้ไฟล์ ' names.dmp ' และ ' nodes.dmp ' ที่ดาวน์โหลดโดยใช้ฟังก์ชั่น --download-taxonomy โดยใช้พารามิเตอร์ --names และ --nodes ตามลำดับ ตารางผลลัพธ์จะมี 10 คอลัมน์ที่ให้ข้อมูลต่อไปนี้:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 : การแก้ไขข้อผิดพลาด -> ปรับปรุงการแยกวิเคราะห์ส่วนหัวที่เป็นตัวหนาระหว่าง --importcrabs --version v 1.0.5 : การแก้ไขข้อผิดพลาด -> เพิ่มข้อจำกัดความยาวให้กับ SEQ ID เมื่อสร้างฐานข้อมูล BLAST ตามต้องการสำหรับซอฟต์แวร์ BLAST+crabs --version v 1.0.4 : เพิ่มข้อมูล-> ให้ข้อมูลที่ถูกต้องเกี่ยวกับอินพุตค่าสำหรับ --pairwise-global-alignment --coverage --percent-identitycrabs --version v 1.0.3 : การแก้ไขข้อผิดพลาด -> ตรวจสอบการตอบสนองของเซิร์ฟเวอร์ NCBI 3 ครั้งก่อนที่จะยกเลิกการวิเคราะห์crabs --version v 1.0.2 : การแก้ไขข้อผิดพลาด -> ความสามารถในการรายงานเมื่อ 0 ลำดับจะถูกส่งคืนหลังจากการวิเคราะห์crabs --version v 1.0.1 : การแก้ไขข้อผิดพลาด -> การสร้างแบบสอบถาม NCBI ที่ประสบความสำเร็จโดยใช้พารามิเตอร์ --species

