nucleotide transformer
1.0.0
ยินดีต้อนรับสู่พื้นที่เก็บข้อมูล InstaDeep Github ซึ่งมีจุดเด่นดังนี้:
เรารู้สึกตื่นเต้นที่ได้ร่วมงานเหล่านี้แบบโอเพ่นซอร์ส และช่วยให้ชุมชนสามารถเข้าถึงโค้ดและน้ำหนักที่ได้รับการฝึกอบรมล่วงหน้าสำหรับแบบจำลองภาษาจีโนมิกส์ทั้งเก้าแบบจำลองและแบบจำลองการแบ่งส่วน 2 แบบ แบบจำลองจากโครงการ nucleotide transformer ได้รับการพัฒนาร่วมกับ Nvidia และ TUM และแบบจำลองดังกล่าวได้รับการฝึกอบรมบนโหนด DGX A100 บน Cambridge-1 แบบจำลองจากโครงการ nucleotide transformer Agro ได้รับการพัฒนาร่วมกับ Google และแบบจำลองที่ได้รับการฝึกเกี่ยวกับเครื่องเร่งปฏิกิริยา TPU-v4
โดยรวมแล้ว งานของเราให้ข้อมูลเชิงลึกใหม่ๆ ที่เกี่ยวข้องกับการฝึกอบรมล่วงหน้าและการประยุกต์ใช้แบบจำลองพื้นฐานภาษา ตลอดจนการฝึกอบรมแบบจำลองที่ใช้เป็นตัวเข้ารหัสแกนหลัก ไปจนถึงจีโนมิกส์ที่มีโอกาสมากมายในการใช้งานในภาคสนาม
ในที่เก็บนี้คุณจะพบสิ่งต่อไปนี้:
เมื่อเปรียบเทียบกับแนวทางอื่นๆ แบบจำลองของเราไม่เพียงแต่บูรณาการข้อมูลจากจีโนมอ้างอิงเดี่ยวเท่านั้น แต่ยังใช้ประโยชน์จากลำดับดีเอ็นเอจากจีโนมมนุษย์ที่หลากหลายกว่า 3,200 รายการ รวมถึงจีโนม 850 รายการจากหลากหลายสายพันธุ์ รวมถึงสิ่งมีชีวิตแบบจำลองและที่ไม่ใช่แบบจำลอง ด้วยการประเมินที่ครอบคลุมและแข็งแกร่ง เราแสดงให้เห็นว่าแบบจำลองขนาดใหญ่เหล่านี้ให้การทำนายฟีโนไทป์ของโมเลกุลที่แม่นยำอย่างยิ่งเมื่อเปรียบเทียบกับวิธีการที่มีอยู่
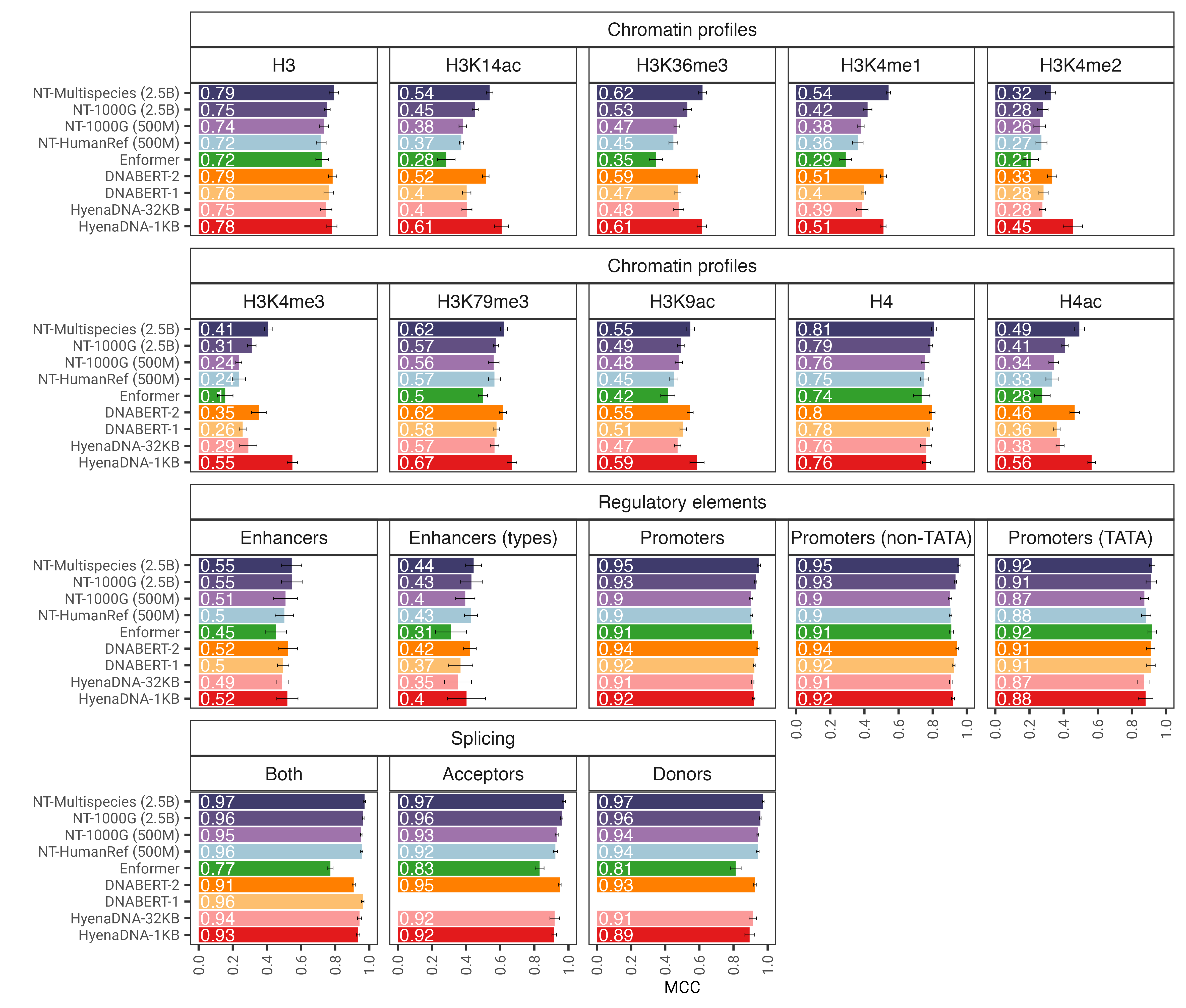
รูปที่ 1: แบบจำลอง nucleotide transformer ทำนายงานจีโนมิกส์ที่หลากหลายได้อย่างแม่นยำหลังจากการปรับแต่งอย่างละเอียด เราแสดงผลการปฏิบัติงานในงานดาวน์สตรีมสำหรับโมเดลหม้อแปลงที่ได้รับการปรับแต่งอย่างละเอียด แถบข้อผิดพลาดแสดงถึง 2 SD ที่ได้มาจากการตรวจสอบข้าม 10 เท่า
ในงานนี้ เรานำเสนอแบบจำลองภาษาขนาดใหญ่ที่เป็นรากฐานแบบใหม่ที่ได้รับการฝึกฝนเกี่ยวกับจีโนมอ้างอิงจากพืช 48 สายพันธุ์โดยเน้นไปที่พันธุ์พืชเป็นหลัก เราประเมินประสิทธิภาพของ AgroNT ในงานทำนายต่างๆ ตั้งแต่คุณสมบัติด้านกฎระเบียบ การประมวลผล RNA และการแสดงออกของยีน และแสดงให้เห็นว่า AgroNT สามารถรับประสิทธิภาพที่ล้ำสมัยได้
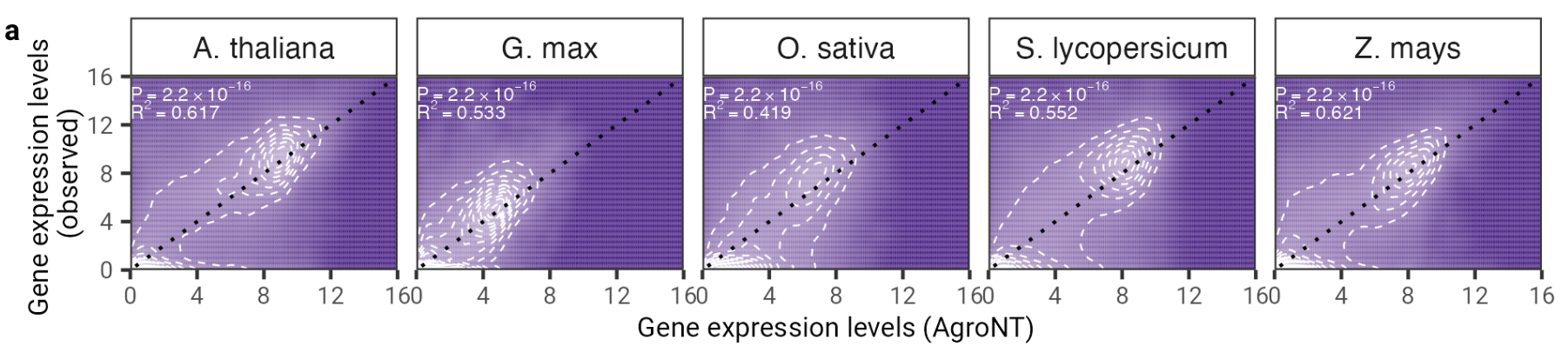
รูปที่ 2: AgroNT แสดงการทำนายการแสดงออกของยีนในพืชชนิดต่างๆ การทำนายการแสดงออกของยีนในยีนที่ค้างอยู่ในเนื้อเยื่อทั้งหมดมีความสัมพันธ์กับระดับการแสดงออกของยีนที่สังเกตได้ ค่าสัมประสิทธิ์การกำหนด (R 2 ) จากแบบจำลองเชิงเส้นและค่า P ที่เกี่ยวข้องระหว่างค่าที่คาดการณ์และค่าที่สังเกตจะแสดงขึ้น
หากต้องการใช้โค้ดและโมเดลที่ได้รับการฝึกล่วงหน้า เพียง:
pip install . -จากนั้น คุณสามารถดาวน์โหลดและทำการอนุมานกับโมเดลทั้งเก้ารุ่นของเราได้โดยใช้โค้ดเพียงไม่กี่บรรทัด:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )ชื่อรุ่นที่รองรับคือ:
คุณยังสามารถรันโมเดลของเราและค้นหาโค้ดตัวอย่างเพิ่มเติมใน google colab
รหัสทำงานทั้งบน GPU และ TPU ต้องขอบคุณ Jax!
เวอร์ชันที่สองของ nucleotide transformer รุ่น v2 ประกอบด้วยชุดของการเปลี่ยนแปลงทางสถาปัตยกรรมที่ได้รับการพิสูจน์แล้วว่ามีประสิทธิภาพมากขึ้น: แทนที่จะใช้การฝังตำแหน่งที่เรียนรู้ เราใช้การฝังแบบหมุนที่ใช้ในแต่ละเลเยอร์ความสนใจและหน่วยเชิงเส้นแบบ Gated พร้อมการเปิดใช้งานแบบ swish โดยไม่มีอคติ โมเดลที่ได้รับการปรับปรุงเหล่านี้ยังยอมรับลำดับโทเค็นสูงสุด 2,048 โทเค็น ซึ่งนำไปสู่หน้าต่างบริบทที่ยาวขึ้นถึง 12kbp ได้รับแรงบันดาลใจจากกฎหมายมาตราส่วน Chinchilla เรายังฝึกอบรมโมเดล NT-v2 ของเราบนชุดข้อมูลหลายสายพันธุ์ของเราสำหรับระยะเวลาที่นานขึ้น (โทเค็น 300B สำหรับรุ่น 50M และ 100M; โทเค็น 1T สำหรับรุ่น 250M และ 500M) เปรียบเทียบกับรุ่น v1 (โทเค็น 300B สำหรับทั้ง 4 รุ่น)
เลเยอร์ของหม้อแปลงเป็นแบบ 1 ดัชนี ซึ่งหมายความว่าการเรียก get_pretrained_model ด้วยอาร์กิวเมนต์ model_name="500M_human_ref" และ embeddings_layers_to_save=(1, 20,) จะส่งผลให้เกิดการแยกการฝังหลังจากเลเยอร์หม้อแปลงตัวแรกและตัวที่ 20 สำหรับหม้อแปลงที่ใช้หัว Roberta LM เป็นเรื่องปกติที่จะแยกการฝังขั้นสุดท้ายหลังจากบรรทัดฐานชั้นแรกของหัว LM แทนที่จะเป็นหลังจากบล็อกหม้อแปลงสุดท้าย ดังนั้น หากเรียกใช้ get_pretrained_model ด้วยอาร์กิวเมนต์ต่อไปนี้ embeddings_layers_to_save=(24,) การฝังจะไม่ถูกแตกหลังจากเลเยอร์หม้อแปลงสุดท้าย แต่จะถูกแยกออกหลังจากบรรทัดฐานของเลเยอร์แรกของส่วนหัว LM
แบบจำลอง SegmentNT ใช้ประโยชน์จาก nucleotide transformer หม้อแปลง (NT) ซึ่งเราถอดส่วนหัวของแบบจำลองภาษาออก และแทนที่ด้วยส่วนหัวของการแบ่งส่วน U-Net 1 มิติ เพื่อทำนายตำแหน่งขององค์ประกอบจีโนมหลายประเภทในลำดับที่ความละเอียดนิวคลีโอไทด์เดียว เรานำเสนอแบบจำลองที่แตกต่างกันสองแบบในองค์ประกอบจีโนมมนุษย์ 14 คลาสที่แตกต่างกันในลำดับอินพุตสูงสุด 30kb สิ่งเหล่านี้รวมถึงยีน (ยีนที่เข้ารหัสโปรตีน, lncRNAs, 5'UTR, 3'UTR, exon, อินตรอน, ตัวรับรอยต่อ และไซต์ผู้บริจาค) และการควบคุม (สัญญาณ polyA, โปรโมเตอร์และเอนแฮนเซอร์ที่ไม่แปรเปลี่ยนของเนื้อเยื่อและเฉพาะเนื้อเยื่อ และขอบเขตของ CTCF เว็บไซต์) องค์ประกอบ SegmentNT บรรลุประสิทธิภาพที่เหนือกว่าสถาปัตยกรรมการแบ่งเซ็กเมนต์ U-Net ที่ล้ำสมัย โดยได้ประโยชน์จากน้ำหนัก NT ที่ได้รับการฝึกอบรมล่วงหน้า และสาธิตภาพรวมแบบ Zero-Shot สูงสุด 50kbp
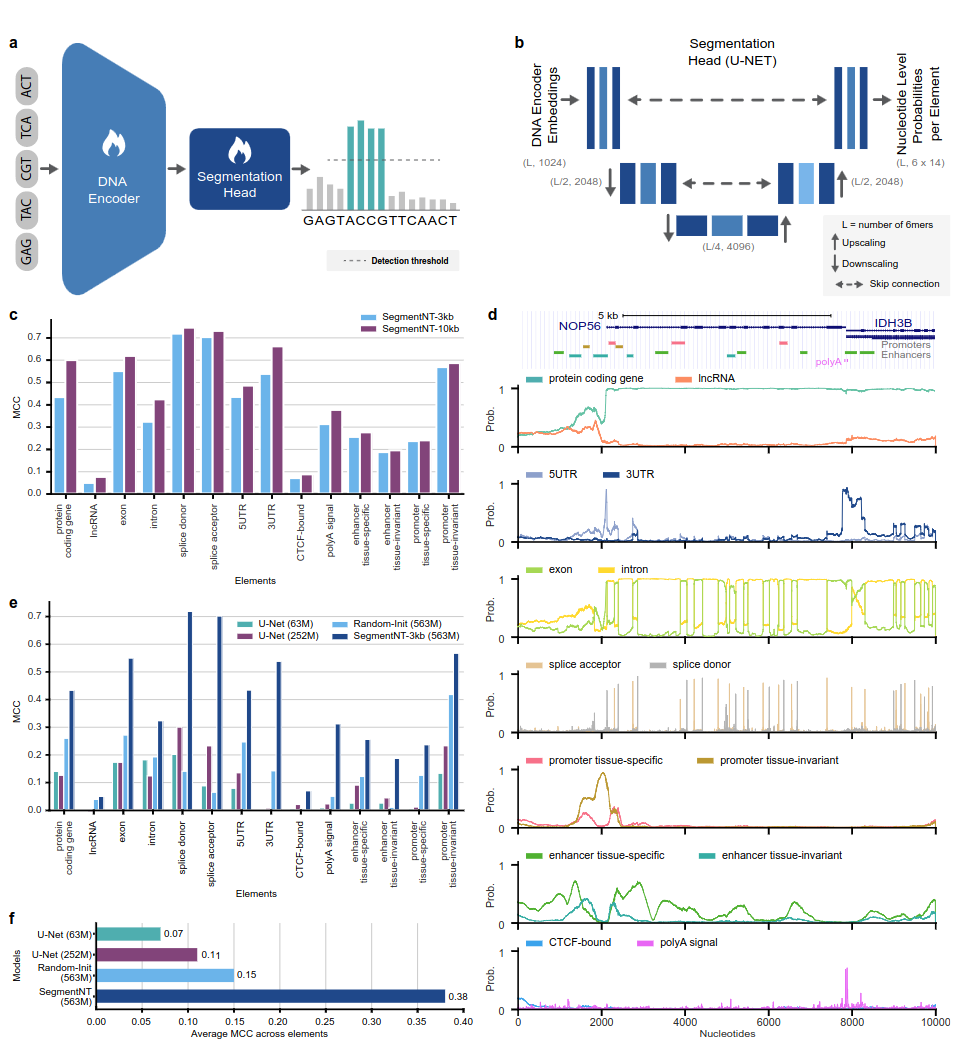
รูปที่ 1: SegmentNT จำกัดวงองค์ประกอบจีโนมที่ความละเอียดนิวคลีโอไทด์
หากต้องการใช้โค้ดและโมเดลที่ได้รับการฝึกล่วงหน้า เพียง:
pip install . -จากนั้น คุณสามารถดาวน์โหลดและอนุมานตามลำดับกับโมเดลใดๆ ของเราโดยใช้โค้ดเพียงไม่กี่บรรทัด:
rescaling factor จะถูกตั้งค่าเป็นปัจจัยที่ใช้ระหว่างการฝึก ในกรณีที่คุณต้องการอนุมานลำดับระหว่าง 30kbp ถึง 50kbp ตรวจสอบให้แน่ใจว่าได้ส่งอาร์กิวเมนต์ rescaling_factor ในฟังก์ชัน get_pretrained_segment_nt_model พร้อมค่า rescaling_factor = max_num_nucleotides / max_num_tokens_nt โดยที่ num_dna_tokens_inference คือจำนวนโทเค็นในการอนุมาน (เช่น 6669 สำหรับลำดับของ 40008 คู่ฐาน) และ max_num_tokens_nt คือจำนวนโทเค็นสูงสุดที่ใช้ฝึกฝนหม้อแปลงนิวคลีโอไทด์ของแกนหลัก เช่น 2048
- examples/inference_segment_nt.ipynb นำเสนอวิธีการอนุมานลำดับขนาด 50kb และวางแผนความน่าจะเป็นในการทำซ้ำรูปที่ 3 ของรายงาน
- โมเดล SegmentNT จะไม่จัดการ "N" ใดๆ ในลำดับอินพุต เนื่องจากแต่ละนิวคลีโอไทด์จำเป็นต้องได้รับโทเค็นเป็น 6-mers ซึ่งไม่สามารถทำได้เมื่อใช้ลำดับที่มีคู่เบส "N" หนึ่งคู่หรือหลายคู่
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )ชื่อรุ่นที่รองรับคือ:
รหัสทำงานทั้งบน GPU และ TPU ต้องขอบคุณ Jax!
โมเดลต่างๆ ได้รับการฝึกฝนเกี่ยวกับลำดับที่มีความยาวสูงสุดถึง 1,000 โทเค็น รวมถึงโทเค็น <CLS> ที่เติมไว้ข้างหน้าโดยอัตโนมัติที่จุดเริ่มต้นของลำดับ tokenizer เริ่มการสร้างโทเค็นจากซ้ายไปขวาโดยการจัดกลุ่มตัวอักษร "A", "C", "G" และ "T" ใน 6 เมอร์ ตัวอักษร "N" ถูกเลือกไม่ให้ถูกจัดกลุ่มภายใน k-mers ดังนั้นเมื่อใดก็ตามที่โทเค็นไนเซอร์พบกับ "N" หรือหากจำนวนนิวคลีโอไทด์ในลำดับไม่เป็นทวีคูณของ 6 มันก็จะทำโทเค็นนิวคลีโอไทด์โดยไม่ต้องจัดกลุ่ม พวกเขา. ตัวอย่างได้รับด้านล่าง:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]ดังนั้นหม้อแปลง v1 และ v2 ทั้งหมดสามารถรับลำดับนิวคลีโอไทด์ได้ถึง 5994 และ 12282 ตามลำดับ หากไม่มี "N" อยู่ข้างใน
คอลเลกชันของแบบจำลองที่นำเสนอในพื้นที่เก็บข้อมูลนี้มีอยู่ในช่องว่างหน้ากอดของ Instadeep ที่นี่: พื้นที่ของ nucleotide transformer และพื้นที่ nucleotide transformer Agro!
เราขอขอบคุณ Maša Roller รวมถึงสมาชิกของ Rostlab โดยเฉพาะ Tobias Olenyi, Ivan Koludarov และ Burkhard Rost สำหรับการอภิปรายอย่างสร้างสรรค์ที่ช่วยระบุทิศทางการวิจัยที่น่าสนใจ นอกจากนี้ เรายังขอขอบคุณทุกคนที่เก็บข้อมูลการทดลองไว้ในฐานข้อมูลสาธารณะ ผู้ที่ดูแลรักษาฐานข้อมูลเหล่านี้ และผู้ที่เผยแพร่วิธีการวิเคราะห์และการคาดการณ์อย่างเสรี เรายังขอขอบคุณทีมพัฒนา Jax
หากคุณพบว่าพื้นที่เก็บข้อมูลนี้มีประโยชน์ในงานของคุณ โปรดเพิ่มข้อมูลอ้างอิงที่เกี่ยวข้องให้กับเอกสารที่เกี่ยวข้องของเรา:
กระดาษ nucleotide transformer :
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}กระดาษ nucleotide transformer เกษตร:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}กระดาษเซกเมนต์ NT
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}หากคุณมีคำถามหรือข้อเสนอแนะเกี่ยวกับโค้ดและรุ่น โปรดติดต่อเรา
ขอขอบคุณที่สนใจงานของเรา!


