ClockstaR
1.0.0

塞巴斯蒂安·杜兴 (Sebastian Duchene)、马丁娜·莫拉克 (Martyna Molak) 和西蒙 YW Ho。
分子生态学、进化和系统发育学 (MEEP) 实验室
生物科学学院
悉尼大学
2015 年 6 月 10 日
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
使用 BSD 距离的导数实现树距离的优化
实现拓扑距离的并行版本
编写拓扑距离聚类教程
集成模型生成器进行模型测试
集成 RaxML 以实现分支长度和拓扑的最大似然优化
使用多基因数据集估计进化时间尺度是系统发育研究中的常见做法。多基因数据集可以按基因、密码子位置或两者进行分区。在本教程中,我们将“数据子集”称为单个基因或多基因数据集的任何子单位。术语“分区”是指一组数据子集。
尽管可以使用单个松弛时钟模型来连接和分析数据子集,但即使它们的树拓扑相同,数据子集之间的谱系间速率变化模式也可能不同。例如,线粒体基因的谱系间比率变异可能与核基因的不同。因此,不同的松弛时钟模型可以分配给不同的数据子集,以改进进化时间尺度和统计拟合的估计(参见 Duchene 和 Ho.,2014a)
多基因数据集的划分方法有很多种。比较分区方案的常见方法是使用贝叶斯因子或基于可能性的标准进行模型拟合。然而,在大多数情况下,测试所有可能的划分方案是不可行的,尤其是使用计算贝叶斯因子的计算密集型方法。
ClockstaR 估计每个数据子集的系统发育分支长度。计算每对树的分支分数距离(称为 sBSDmin),作为其谱系间比率变异模式差异的度量。这些距离用于使用 GAP 统计数据和 PAM 聚类算法来推断最佳分区策略,如在包 cluster 中实现的那样 (Maechler et al., 2012)(有关 sBSDmin 指标的详细信息,请参阅 Duchene et al., 2014b) 。
ClockstaR 是一个 R 软件包,用于多基因数据集的系统发育分子时钟分析。它使用不同基因的谱系率变异模式来选择时钟分区策略。该方法使用系统发育树距离度量和无监督机器学习算法来确定时钟分区的最佳数量,以及应在每个分区下分析哪些基因。 ClocsktaR 中选择的分区策略可用于后续的分子钟分析,例如 BEAST、MrBayes、PhyloBayes 等程序。
请点击此链接查看原始出版物。
ClockstaR 需要安装 R。它还需要一些 R 依赖项,这些依赖项可以通过 R 获得,如下所述。
请将任何请求或问题发送至 Sebastian Duchene (sebastian.duchene[at]sydney.edu.au)。其他一些软件和资源可以在悉尼大学的分子生态学、进化和系统发育实验室找到。
下载此存储库作为 zip 文件并解压缩。以下说明使用clockstar_example_data文件夹,其中包含一些fasta文件和newick格式的系统发育树。在文本编辑器(例如 text wrangler)中打开这些文件中的任何一个。这些数据是在四种进化速率变化模式下模拟的。请注意,树是所有基因或数据分区的树拓扑。要运行 ClockstaR,请格式化您的数据,类似于 Clockstar_example_data 中的示例数据。
ClockstaR 可以直接从 GitHub 安装。这需要 devtools 包。在 R 提示符处键入以下代码以安装所有必要的工具(请注意,您需要互联网连接才能直接下载软件包):。
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )下载并安装后,加载ClockstaR的函数库。
library (ClockstaR2)要查看程序如何运行的示例,请输入:
example (ClockstaR2)本教程的其余部分使用 Clockstar_example_data 文件夹
第一步是获取每个比对的基因树。为此,我们使用树拓扑,并使用每个单独的基因比对(在本例中为 A1.fasta 到 C3.fasta)优化分支长度。如果您有基因树,则以 newick 格式保存在文件中,然后进入下一步(交互式运行 Clockstar)。
在 R 提示符中输入以下代码并按 Enter 键:
optim . trees . interactive ()如果您收到有关安装 phangorn 包的错误消息,请使用此代码,然后重复 optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR 将打印以下消息:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICK将 Clockstar_example_data 文件夹拖到 R 控制台并输入 Enter。请注意,该文件夹应仅包含 FASTA 格式的排列和 NEWICK 格式的树形拓扑。您将看到以下消息:
What should be the name of the file to save the optimised trees ?输入优化树的文件名。在这种情况下,我们将使用“example.trees”
example . trees此时,ClockstaR 会询问是否应该对每个基因使用单独的替换模型,或者在所有情况下都使用 JC。由于这些数据是在 JC 下模拟的,因此我们将键入“n”并按 Enter 键。键入“y”以分别指定每个替代模型。
输入“n”并按 Enter 键后,ClockstaR 将开始运行。它将在图形设备中打印基因树。如果指定的树已扎根,它还可能会打印一些警告,可以安全地忽略这些警告。
打开clockstar_example_data 文件夹。您将找到一个名为“example.trees”的文件,如上面几个步骤所指定。在文本编辑器中打开 example.trees。它包含每个基因树和根据基因比对名称的树名称。它应该看起来像这样:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.该包含树的文件将用于下一步。
对于此步骤,必须将基因树保存在文件中,例如在上一步中获得的文件。
打开 R 并加载 ClockstaR,如上所示。在提示符下键入以下代码:
clockstar . interactive ()ClockstaR 将打印以下消息:
please drag or type in the path to your gene trees file in NEWICK format :将带有基因树的文件拖到 R 控制台。如果您按照上一步操作,该文件将被称为 example.trees。输入回车。
根据您安装的软件包,ClockstaR 可能会询问是否应该并行运行。这对于大型数据集非常有效。但对于示例数据来说,它不会产生很大的差异,因此如果您看到此消息,请输入“n”,然后输入 Enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar 现在将开始运行。屏幕上的输出应如下所示:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.估计树距离(在原始出版物中描述)后,ClockstaR 将打印以下消息:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)这些是聚类算法的设置。它们适用于大多数数据集,因此在本例中我们可以键入“y”,然后输入。通过键入“n”我们可以更改这些设置,有关更多详细信息,请参阅 Kaufman 和 Rousseeuw (2009)。
ClockstaR 现在将运行聚类算法。最后它会打印最佳分区数并询问是否应将结果保存在 pdf 文件中:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)输入“y”然后回车。
然后 ClockstaR 将询问输出文件的名称:
What should be the name and path of the output file ?对于本示例,键入“example_run”并回车,但可以使用任何名称。
现在打开clockstar_example_data文件夹并打开两个pdf文件example_run_gapstats.pdf和example_run_matrix.pdf。
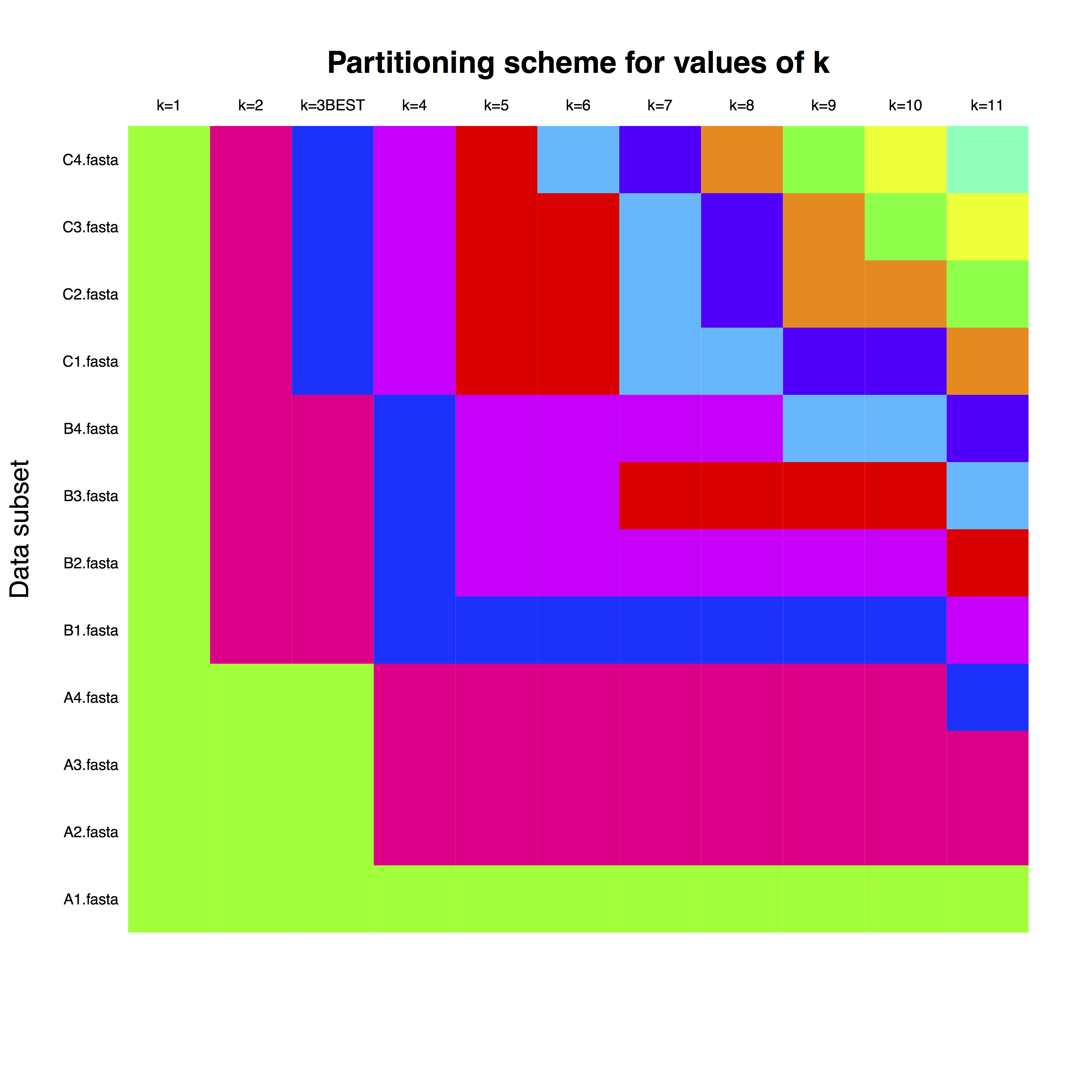
example_run_matrix 是矩阵,其中行对应于每个基因,如 FASTA 文件中所命名。列是分区的数量,颜色代表每个基因对时钟分区的分配。例如,对于k = 3(这是最佳分区数),可以对具有字母 A、B 和 C 的基因使用单独的时钟分区。
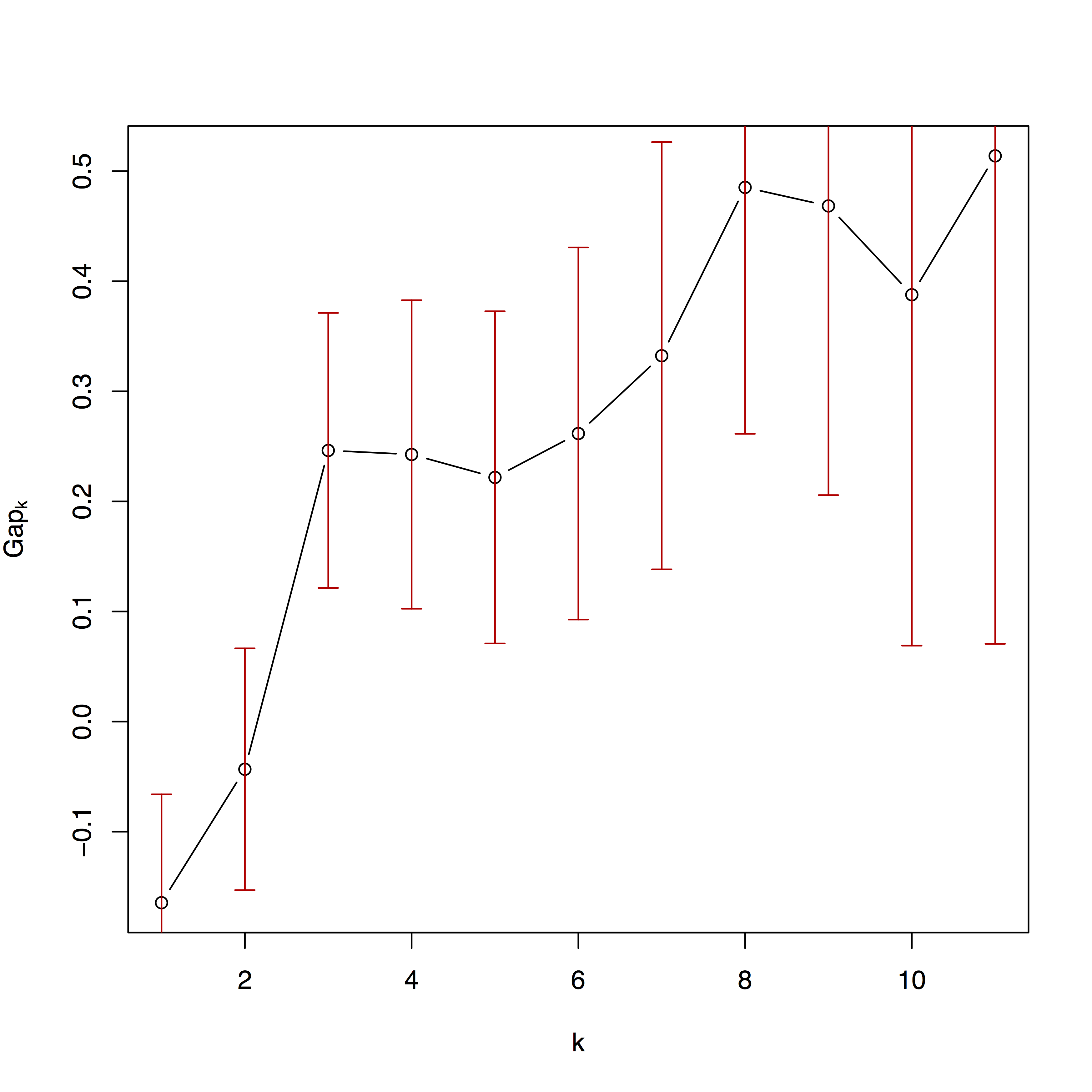
第二张图是聚类算法在不同数量的分区上的拟合情况。更多详细信息请参阅 Kaufman 和 Rousseeuw (2009) 以及包 cluster 的文档。
ClockstaR 可以使用其他自定义设置运行。请参阅文档了解其他详细信息,如果有任何问题,请致电 sebastian.duchene[at]sydney.edy.au。
标志由童俊设计
Duchene, S. 和 Ho, SY (2014a)。使用多个松弛时钟模型根据 DNA 序列数据估计进化时间尺度。分子系统发育学和进化(77):65-70。
Duchene, S.、Molak, M. 和 Ho, SY (2014b)。 ClockstaR:在分子系统发育分析中选择松弛时钟模型的数量。生物信息学30 (7): 1017-1019。
考夫曼,L.,和卢梭,PJ (2009)。查找数据中的组:聚类分析简介(第 344 卷)。约翰·威利父子。


