该仓库包含:
sepal需要python3 ,最好是 3.5 以上的版本。要下载并安装,请打开终端并更改为您想要将sepal下载到的目录,然后执行以下操作:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
根据您的用户权限,您可能需要添加--user作为setup.py的参数。运行该设置将为您提供计算扩散时间所需的最少安装。但是,如果您希望能够使用分析模块,您还需要安装推荐的软件包。为此,只需(在同一目录中)运行:
pip install -e " .[full] "同样,可能需要包含--user 。另外,如果这是您设置python-pip接口的方式,您可能必须使用pip3 。如果您使用conda或虚拟环境,请按照他们的建议安装软件包。
这应该安装命令行界面 (CLI) 和标准包。要测试并查看安装是否成功,您可以尝试执行以下命令:
sepal -h
它应该打印与萼片相关的帮助消息。如果到目前为止一切顺利,您可以继续查看示例部分以查看sepal实际应用!
建议通过命令行界面使用 sepal。计算扩散时间的模拟以及随后的结果分析或检查都可以通过键入sepal并随后run或analyze来轻松执行。 analyze模块有不同的选项,用于可视化结果( inspect )、将配置文件分类到模式家族( family )或对已识别的家族进行功能富集分析( fea )。有关可用命令的完整列表,请执行sepal module -h ,其中 module 是run和analyze之一。下面,我们说明如何使用萼片来查找具有空间模式的转录谱。
我们将创建一个文件夹来保存我们的结果,该文件夹也将作为我们的工作目录。从存储库的主目录中,执行以下操作:
cd res
mkdir example
cd exampleMOB 样本将用于举例说明我们的分析。我们首先计算每个转录谱的扩散时间:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1下面是一个示例(附加显示了帮助命令),展示了它的外观

计算出扩散时间后,我们想要检查结果,就像在研究中一样,我们将查看前 20 个配置文件。我们可以通过运行以下命令轻松地根据结果生成图像:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5这看起来像这样:

输出将如下图所示:
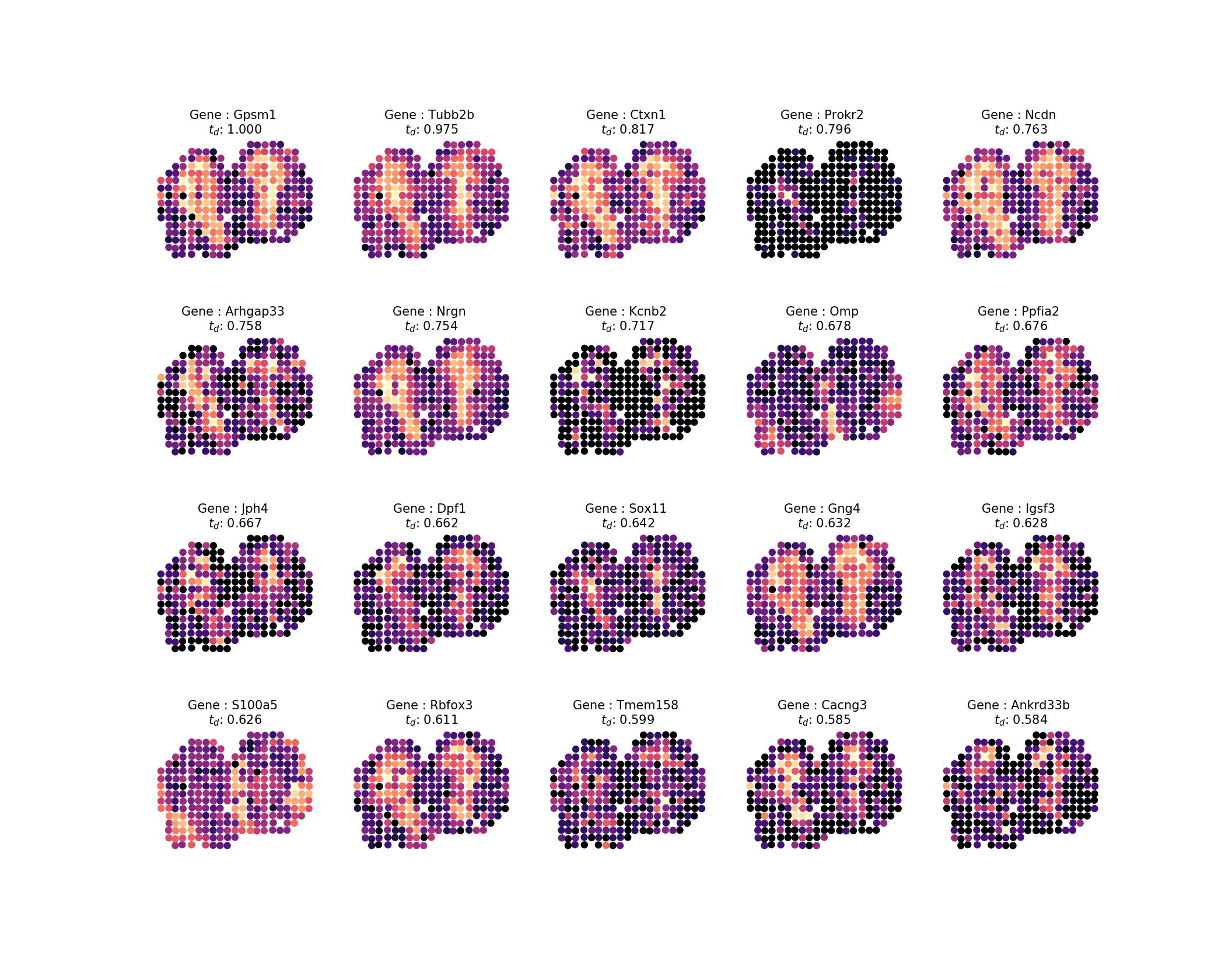
然后,要将排名前 100 的基因分类到一组模式族中,其中模式中 85% 的方差应由特征模式来解释,请执行以下操作:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3由此,我们得出每个家族的以下三个代表性主题:
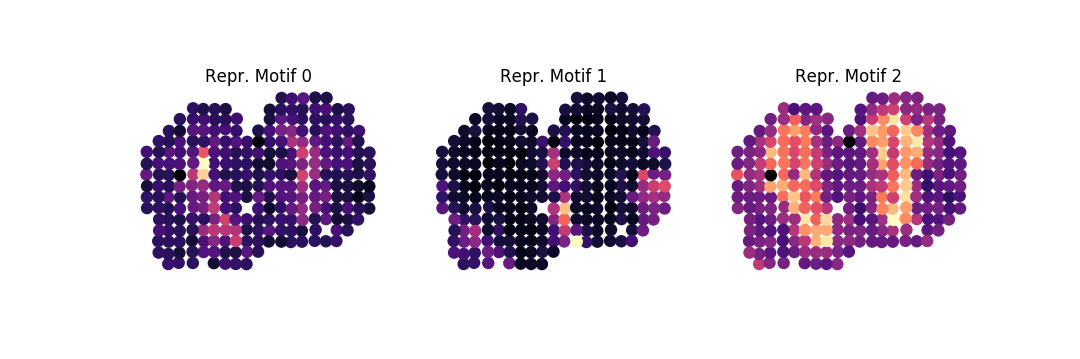
我们可以通过运行以下命令对我们的家庭进行富集分析:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "例如,我们看到 Family 2 丰富了与神经元功能、生成和调节相关的几个过程:
| 家庭 | 本国的 | 姓名 | p_值 | 来源 | 交叉点大小 | |
|---|---|---|---|---|---|---|
| 2 | 2 | GO:0007399 | 神经系统发育 | 0.00035977 | 去:BP | 26 |
| 3 | 2 | 号码:0050773 | 树突发育的调控 | 0.000835883 | 去:BP | 8 |
| 4 | 2 | GO:0048167 | 突触可塑性的调节 | 0.00196494 | 去:BP | 8 |
| 5 | 2 | 号码:0016358 | 枝晶发育 | 0.00217167 | 去:BP | 9 |
| 6 | 2 | 号码:0048813 | 树突形态发生 | 0.00741589 | 去:BP | 7 |
| 7 | 2 | 号码:0048814 | 树突形态发生的调控 | 0.00800399 | 去:BP | 6 |
| 8 | 2 | 号码:0048666 | 神经元发育 | 0.0114088 | 去:BP | 16 |
| 9 | 2 | 号码:0099004 | 钙调蛋白依赖性激酶信号通路 | 0.0159572 | 去:BP | 3 |
| 10 | 2 | GO:0050804 | 化学突触传递的调节 | 0.0341913 | 去:BP | 10 |
| 11 | 2 | GO:0099177 | 跨突触信号传导的调节 | 0.0347783 | 去:BP | 10 |
当然,这种分析绝不是详尽无遗的。而是一个简单的示例来展示如何操作sepal的 CLI。
虽然sepal被设计为独立工具,但我们还将其构建为标准 python 包,可以从中导入函数并在集成工作流程中使用。为了说明如何做到这一点,我们提供了一个示例,重现黑色素瘤分析。稍后可能会添加更多示例。
sepal的输入必须采用n_locations x n_genes格式,但是如果您的数据以相反的方式构建( n_genes x n_locations ),则只需在运行模拟或分析时提供--transpose标志即可的。
我们目前支持.csv 、 .tsv和.h5ad格式。对于后者,您的文件应根据此格式构建。我们预计scanpy团队将在不久的将来发布一个版本,其中提出了空间数据的标准化格式,但在那之前我们将使用上述标准。
我们使用的所有真实数据都是公开的,可以通过以下链接找到:
合成数据是通过以下方式生成的:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py 研究中提出的所有结果都可以在res文件夹中找到,包括真实数据和合成数据。对于每个样本,我们都相应地构建了结果:
res/sample-name/X-diffusion-times.tsv :所有排序基因的扩散时间analysis/ :包含二次分析的输出

