nucleotide transformer
1.0.0
欢迎来到 InstaDeep Github 存储库,其中有:
我们很高兴能够开源这些作品,并为社区提供这九种基因组语言模型和 2 个分割模型的代码和预训练权重的访问权限。 nucleotide transformer项目的模型是与 Nvidia 和 TUM 合作开发的,并且这些模型在 Cambridge-1 上的 DGX A100 节点上进行训练。 Agro nucleotide transformer项目的模型是与 Google 合作开发的,并在 TPU-v4 加速器上进行了训练。
总的来说,我们的工作提供了与语言基础模型的预训练和应用相关的新颖见解,以及使用它们作为骨干编码器的模型的训练,以及基因组学在该领域的应用的充足机会。
在此存储库中,您将找到以下内容:
与其他方法相比,我们的模型不仅整合了来自单个参考基因组的信息,还利用了来自 3,200 多个不同人类基因组的 DNA 序列,以及来自广泛物种(包括模型和非模型生物)的 850 个基因组。通过稳健和广泛的评估,我们表明,与现有方法相比,这些大型模型提供了极其准确的分子表型预测。
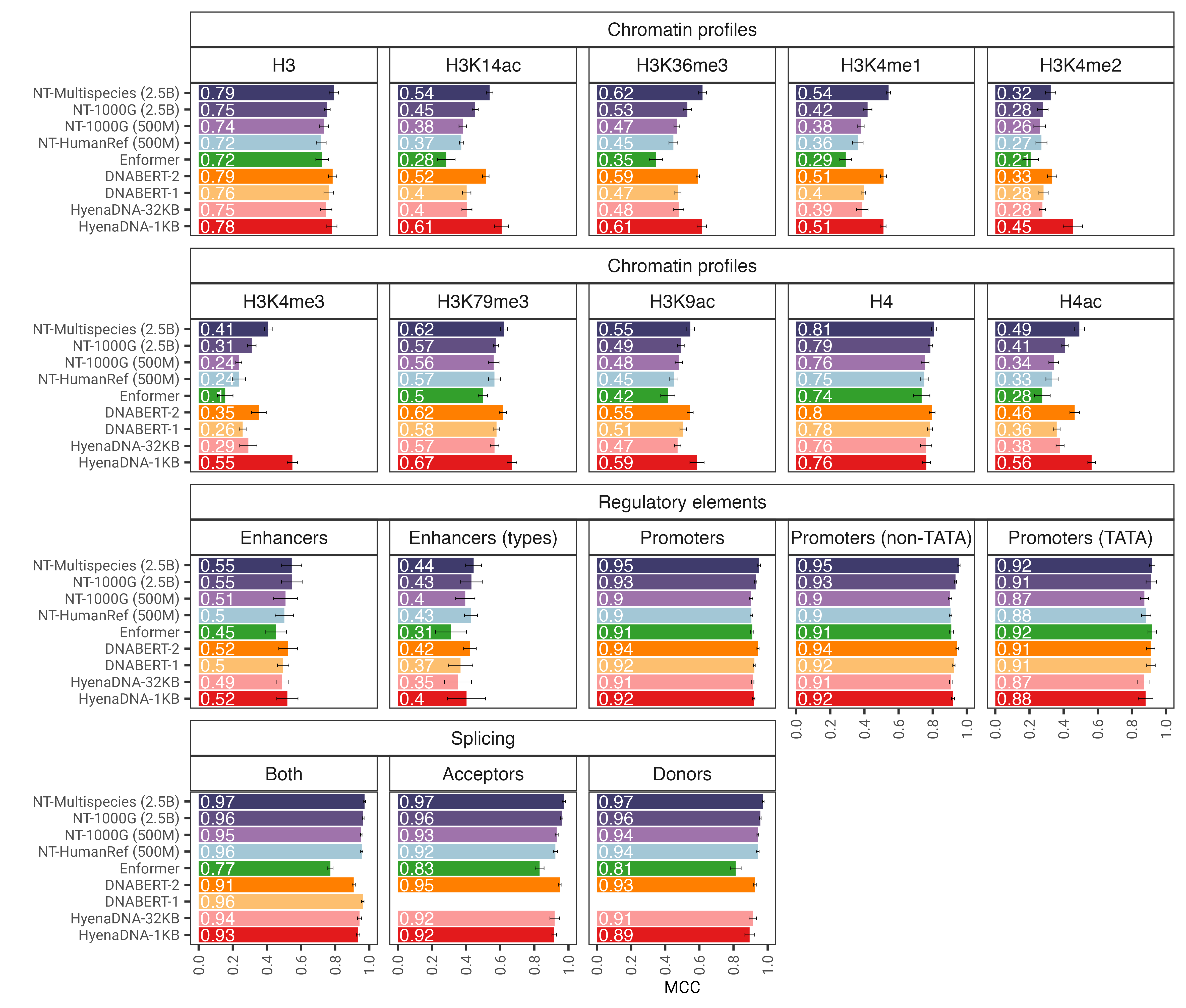
图 1:经过微调后, nucleotide transformer模型可以准确预测各种基因组学任务。我们展示了微调变压器模型的下游任务的性能结果。误差线代表源自 10 倍交叉验证的 2 个 SD。
在这项工作中,我们提出了一种新颖的基础大语言模型,该模型在 48 种植物物种的参考基因组上进行训练,主要关注作物物种。我们评估了 AgroNT 在调控特征、RNA 处理和基因表达等多个预测任务中的性能,并表明 AgroNT 可以获得最先进的性能。
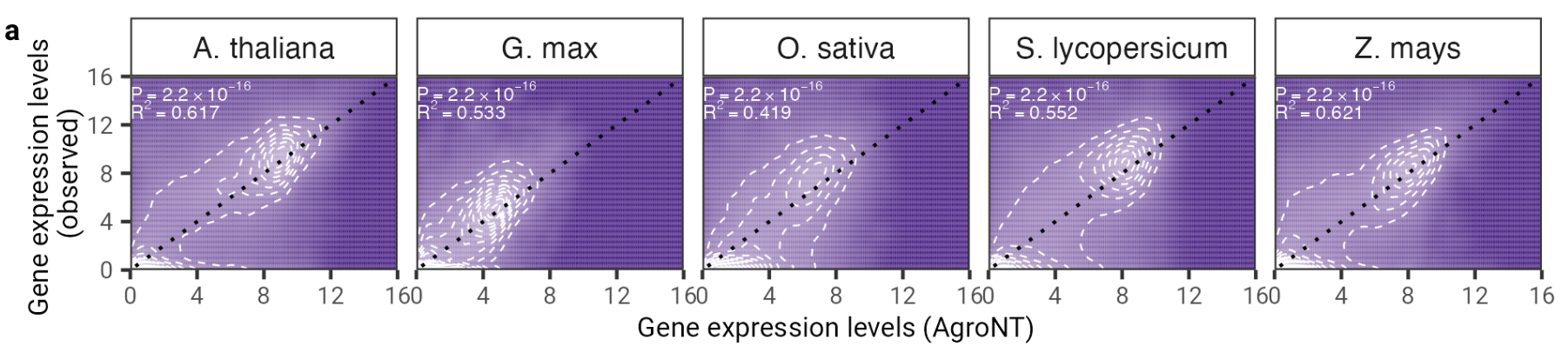
图 2:AgroNT 提供不同植物物种的基因表达预测。所有组织中保留基因的基因表达预测与观察到的基因表达水平相关。显示了线性模型的确定系数 (R 2 ) 以及预测值和观测值之间的相关 P 值。
要使用代码和预训练模型,只需:
pip install . 。然后,您只需几行代码即可下载并使用我们的九个模型中的任何一个进行推理:
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_model
# Get pretrained model
parameters , forward_fn , tokenizer , config = get_pretrained_model (
model_name = "500M_human_ref" ,
embeddings_layers_to_save = ( 20 ,),
max_positions = 32 ,
)
forward_fn = hk . transform ( forward_fn )
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCG" , "ATTTCTCTCTCTCTCTGAGATCGATCGATCGAT" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
# Initialize random key
random_key = jax . random . PRNGKey ( 0 )
# Infer
outs = forward_fn . apply ( parameters , random_key , tokens )
# Get embeddings at layer 20
print ( outs [ "embeddings_20" ]. shape )支持的型号名称有:
您还可以运行我们的模型并在 google colab 中找到更多示例代码
感谢 Jax,代码可以在 GPU 和 TPU 上运行!
我们的第二个版本的nucleotide transformer v2 模型包括一系列被证明更有效的架构变化:我们不使用学习的位置嵌入,而是使用每个注意层使用的旋转嵌入和具有无偏差的快速激活的门控线性单元。这些改进的模型还接受多达 2,048 个标记的序列,从而产生 12kbp 的更长上下文窗口。受 Chinchilla 缩放定律的启发,与 v1 模型(300B 令牌)相比,我们还在多物种数据集上训练 NT-v2 模型,训练持续时间更长(50M 和 100M 模型使用 300B 令牌;250M 和 500M 模型使用 1T 令牌)适用于所有四种型号)。
转换器层的索引为 1,这意味着使用参数model_name="500M_human_ref"和embeddings_layers_to_save=(1, 20,)调用get_pretrained_model将导致在第一个和第 20 个转换器层之后提取嵌入。对于使用 Roberta LM head 的 Transformer,通常的做法是在 LM head 的第一层范数之后而不是在最后一个 Transformer 块之后提取最终嵌入。因此,如果使用以下参数调用get_pretrained_model embeddings_layers_to_save=(24,) ,则嵌入不会在最终 Transformer 层之后提取,而是在 LM 头的第一层范数之后提取。
SegmentNT 模型利用nucleotide transformer (NT) 变换器,我们从中删除了语言模型头并替换为一维 U-Net 分段头,以单核苷酸分辨率预测序列中几种类型的基因组元素的位置。我们在高达 30kb 的输入序列中针对 14 种不同类别的人类基因组元素提出了两种不同的模型变体。这些包括基因(蛋白质编码基因、lncRNA、5'UTR、3'UTR、外显子、内含子、剪接受体和供体位点)和调节(polyA 信号、组织不变和组织特异性启动子和增强子,以及 CTCF 结合站点)元素。受益于 NT 的预训练权重,SegmentNT 实现了优于最先进的 U-Net 分割架构的性能,并展示了高达 50kbp 的零样本泛化能力。
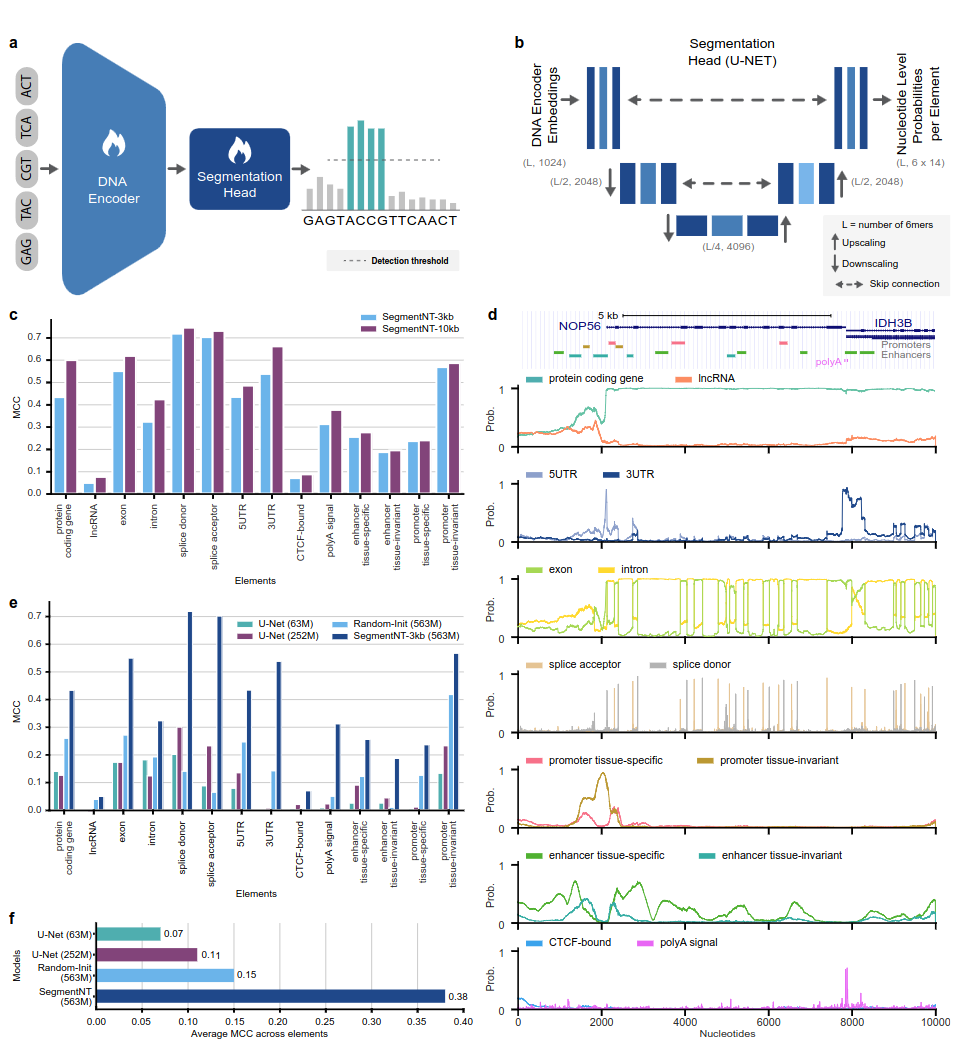
图 1:SegmentNT 以核苷酸分辨率定位基因组元件。
要使用代码和预训练模型,只需:
pip install . 。然后,您只需几行代码即可下载并使用我们的任何模型推断序列:
rescaling factor设置为训练期间使用的因子。如果您需要推断 30kbp 到 50kbp 之间的序列,请确保在get_pretrained_segment_nt_model函数中传递rescaling_factor参数,其值为rescaling_factor = max_num_nucleotides / max_num_tokens_nt其中num_dna_tokens_inference是推断时的标记数(即 6669 对于 40008 个碱基的序列)对)和max_num_tokens_nt是骨干核苷酸转换器训练的最大标记数,即2048 。
?笔记本examples/inference_segment_nt.ipynb展示了如何推断 50kb 序列并绘制概率以重现论文的图 3。
? SegmentNT 模型不处理输入序列中的任何“N”,因为每个核苷酸都需要标记为 6 聚体,而使用包含一个或多个“N”碱基对的序列时则不能出现这种情况。
import haiku as hk
import jax
import jax . numpy as jnp
from nucleotide_transformer . pretrained import get_pretrained_segment_nt_model
# Initialize CPU as default JAX device. This makes the code robust to memory leakage on
# the devices.
jax . config . update ( "jax_platform_name" , "cpu" )
backend = "cpu"
devices = jax . devices ( backend )
num_devices = len ( devices )
print ( f"Devices found: { devices } " )
# The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by
# 2 to the power of the number of downsampling block, i.e 4.
max_num_nucleotides = 8
assert max_num_nucleotides % 4 == 0 , (
"The number of DNA tokens (excluding the CLS token prepended) needs to be dividible by"
"2 to the power of the number of downsampling block, i.e 4." )
parameters , forward_fn , tokenizer , config = get_pretrained_segment_nt_model (
model_name = "segment_nt" ,
embeddings_layers_to_save = ( 29 ,),
attention_maps_to_save = (( 1 , 4 ), ( 7 , 10 )),
max_positions = max_num_nucleotides + 1 ,
)
forward_fn = hk . transform ( forward_fn )
apply_fn = jax . pmap ( forward_fn . apply , devices = devices , donate_argnums = ( 0 ,))
# Get data and tokenize it
sequences = [ "ATTCCGATTCCGATTCCAACGGATTATTCCGATTAACCGATTCCAATT" , "ATTTCTCTCTCTCTCTGAGATCGATGATTTCTCTCTCATCGAACTATG" ]
tokens_ids = [ b [ 1 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens_str = [ b [ 0 ] for b in tokenizer . batch_tokenize ( sequences )]
tokens = jnp . asarray ( tokens_ids , dtype = jnp . int32 )
random_key = jax . random . PRNGKey ( seed = 0 )
keys = jax . device_put_replicated ( random_key , devices = devices )
parameters = jax . device_put_replicated ( parameters , devices = devices )
tokens = jax . device_put_replicated ( tokens , devices = devices )
# Infer on the sequence
outs = apply_fn ( parameters , keys , tokens )
# Obtain the logits over the genomic features
logits = outs [ "logits" ]
# Transform them in probabilities
probabilities = jnp . asarray ( jax . nn . softmax ( logits , axis = - 1 ))[..., - 1 ]
print ( f"Probabilities shape: { probabilities . shape } " )
print ( f"Features inferred: { config . features } " )
# Get probabilities associated with intron
idx_intron = config . features . index ( "intron" )
probabilities_intron = probabilities [..., idx_intron ]
print ( f"Intron probabilities shape: { probabilities_intron . shape } " )支持的型号名称有:
感谢 Jax,代码可以在 GPU 和 TPU 上运行!
这些模型在长度最多 1000 个标记的序列上进行训练,包括自动添加到序列开头的 <CLS> 标记。分词器通过将字母“A”、“C”、“G”和“T”分组为 6 聚体,开始从左到右分词。 “N”字母被选择不分组在 k-mers 内,因此每当分词器遇到“N”时,或者如果序列中的核苷酸数量不是 6 的倍数,它将对核苷酸进行分词而不分组他们。举例如下:
dna_sequence_1 = "ACGTGTACGTGCACGGACGACTAGTCAGCA"
tokenized_dna_sequence_1 = [ < CLS > , < ACGTGT > , < ACGTGC > , < ACGGAC > , < GACTAG > , < TCAGCA > ]
dna_sequence_2 = "ACGTGTACNTGCACGGANCGACTAGTCTGA"
tokenized_dna_sequence_2 = [ < CLS > , < ACGTGT > , < A > , < C > , < N > , < TGCACG > , < G > , < A > , < N > , < CGACTA > , < GTCTGA > ]因此,如果内部没有“N”,所有 v1 和 v2 转化子可以分别采用最多 5994 和 12282 个核苷酸的序列。
该存储库中提供的模型集合可在 Instadeep 的拥抱空间中找到: nucleotide transformer空间和农业nucleotide transformer空间!
我们感谢 Maša Roller 以及 Rostlab 的成员,特别是 Tobias Olenyi、Ivan Koludarov 和 Burkhard Rost 的建设性讨论,帮助确定了有趣的研究方向。此外,我们向所有将实验数据存入公共数据库的人、维护这些数据库的人以及免费提供分析和预测方法的人表示感谢。我们还感谢 Jax 开发团队。
如果您发现此存储库对您的工作有用,请在我们的相关论文中添加相关引用:
nucleotide transformer论文:
@article { dalla2023nucleotide ,
title = { The nucleotide transformer : Building and Evaluating Robust Foundation Models for Human Genomics } ,
author = { Dalla-Torre, Hugo and Gonzalez, Liam and Mendoza Revilla, Javier and Lopez Carranza, Nicolas and Henryk Grywaczewski, Adam and Oteri, Francesco and Dallago, Christian and Trop, Evan and Sirelkhatim, Hassan and Richard, Guillaume and others } ,
journal = { bioRxiv } ,
pages = { 2023--01 } ,
year = { 2023 } ,
publisher = { Cold Spring Harbor Laboratory }
}农业nucleotide transformer纸:
@article { mendoza2024foundational ,
title = { A foundational large language model for edible plant genomes } ,
author = { Mendoza-Revilla, Javier and Trop, Evan and Gonzalez, Liam and Roller, Ma{v{s}}a and Dalla-Torre, Hugo and de Almeida, Bernardo P and Richard, Guillaume and Caton, Jonathan and Lopez Carranza, Nicolas and Skwark, Marcin and others } ,
journal = { Communications Biology } ,
volume = { 7 } ,
number = { 1 } ,
pages = { 835 } ,
year = { 2024 } ,
publisher = { Nature Publishing Group UK London }
}段NT纸
@article { de2024segmentnt ,
title = { SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models } ,
author = { de Almeida, Bernardo P and Dalla-Torre, Hugo and Richard, Guillaume and Blum, Christopher and Hexemer, Lorenz and Gelard, Maxence and Pandey, Priyanka and Laurent, Stefan and Laterre, Alexandre and Lang, Maren and others } ,
journal = { bioRxiv } ,
pages = { 2024--03 } ,
year = { 2024 } ,
publisher = { Cold Spring Harbor Laboratory }
}如果您对代码和模型有任何疑问或反馈,请随时与我们联系。
感谢您对我们工作的兴趣!


