poseidon
1.0.3

请注意,Poseidon的代码刚刚转移到NextFlow,因此可能仍然有一些错误。请随时报告问题!
在这里,我们提出了Poseidon,这是一种在多个蛋白质编码序列的比对中检测出明显阳性选择位点的管道和可能的重组事件。经历积极选择的地点为您的序列的进化史提供了见解,例如显示重要的突变热点,这是在进化过程中作为病毒宿主武器种族的结果积累的。
波塞冬依靠各种不同的第三方工具(见下文)。但是不用担心,我们将每个工具封装在其自己的Docker容器中,并将它们连接到Workflow Management System NextFlow中。
与Zhou等人的最新研究相比,与SARS-COV-2尖峰蛋白的Poseidon输出的一个小例子。 2020。
您只需要安装的NextFlow(版本20.+)和Docker即可运行管道。所有依赖项将自动拉动。
要么通过克隆这个存储库来运行波塞冬:
git克隆https://github.com/hoelzer/poseidon.gitcd poseidon nextflow运行poseidon.nf------
或让NextFlow进行拉动
NextFlow拉动Hoelzer/Poseidon
我们建议使用特定版本的Poseidon通过
#pullnextflow拉力hoelzer/poseidon -r v1.0.1#runnextflow run hoelzer/poseidon -r v1.0.1---------
根据您的安装步骤,请通过git pull或nextflow pull hoelzer/poseidon更新管道。
重要的是:波塞冬需要以正确的开放阅读框为输入的核苷酸序列。此外,结果在很大程度上取决于您的序列选择,因此,您可以考虑多次以不同的输入序列采样来运行管道。此外,管道无法与太多序列一起使用,因为在其核心Poseidon中,使用了未针对> 100个序列的PAML Suite的Codeml。请在下面找到输入参数和设置的详细说明。
NextFlow可以在您的本地计算机,高性能集群或云等不同环境上轻松执行。不同的-profile用于判断NextFlow应该使用哪个系统。对于本地执行-profile local,docker (也是默认值)。您还可以通过奇异性通过-profile lsf,singularity , -profile slurm,singularity或-profile sge,singularity在HPC上运行Poseidon。在这种情况下,还请考虑调整--cachedir涉及将奇点图像存储在群集上的位置。参数--workdir可能也有助于调整存储临时工作目录的位置(例如,使用/scratch而不是/tmp取决于您的HPC配置。)
现在,假设您使用NextFlow来拉动Poseidon代码,然后使用默认配置文件-profile local,docker在本地计算机上执行管道。
#show help nextflow run hoelzer/poseidon-----##在带有#的本地计算机上运行小示例(首次需要更多时间,因为下载了码头容器)nextflow run hoelzer/poseidon -r v1.0.1 -fasta -fasta 〜/.nextflow/Assets/hoelzer/poseidon/test_data/bats_mx1_small.fasta - 核4#恢复破裂的Runnextflow Run Hoelzer/poseidon -r v1.0.1 -fasta〜/.nextflow/.nextflow/assets/hoelzer/hoelzer/poseidon/test_data/bats_mx1_small.fasta - 核4-库#,而不是使用所有可用的内核,仅在本地MachineNextflow运行Hoelzer/Poseidon -r v1.0.1 -fasta〜/.nextflow/assets/hoelzer/hoelzer/poseidon/test_data/bats_mx1_small.fasta.small.fasta -max_cores 8 - 核4
为了再现Fuchs等人报告的阳性选择结果。 (2017),病毒学杂志运行:
NextFlow Run Hoelzer/Poseidon -R V1.0.0 -Fasta〜/.NextFlow/Assets/hoelzer/hoelzer/poseidon/test_data/bats_mx1.fasta - 核4 -KH-OUTGroup“ Pteropus_alecto,eidolon_helvum,rousettus_aegyptiacus,hypsignatus_monstrosus” -Reference“ myotis_daubentonii”
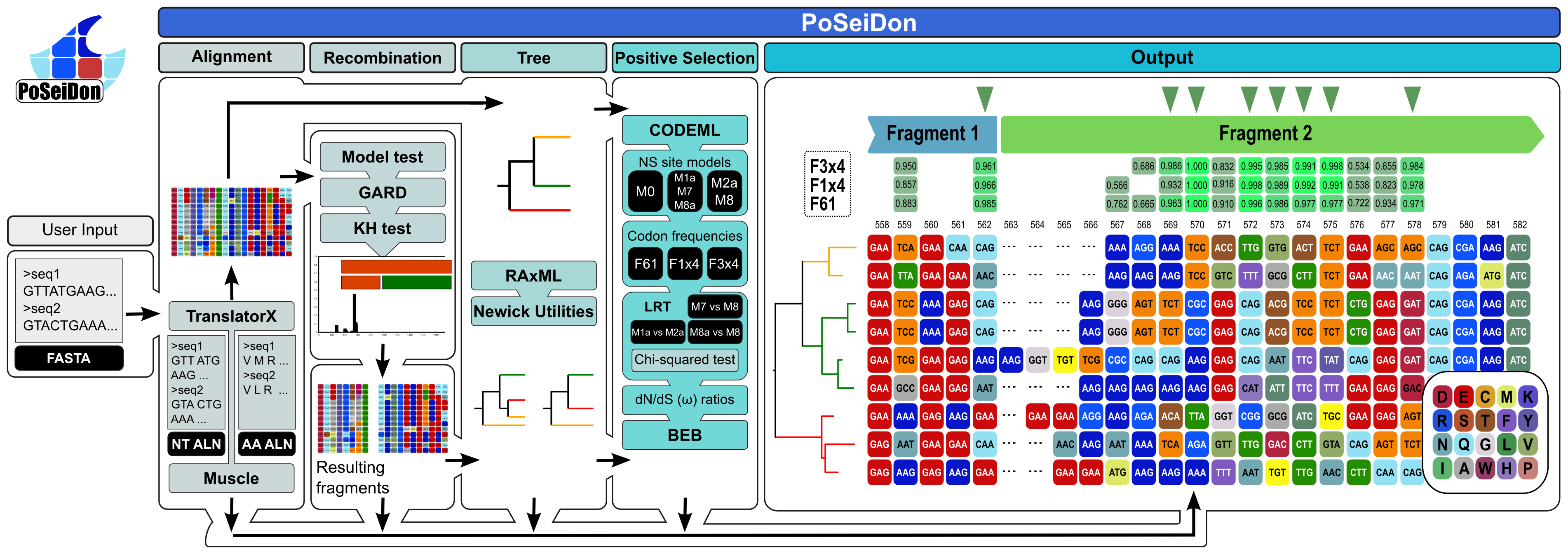
Poseidon管道包括同源蛋白质编码序列的框架对齐,检测假定的重组事件和进化断点,系统发育重建以及在完整对齐中的正面选择位点的检测以及所有可能的片段。最后,在用户友好且清晰的HTML网页中将所有结果组合和可视化。所得的比对片段在HTML输出中用彩色条表示。
Translatorx(V1.1),Abascal等。 (2010); 20435676
肌肉(v3.8.31),埃德加(2004); 15034147
RAXML(V8.0.25),Stamatakis(2014); 24451623
Newick Utilities(V1.6),Junier和Zdobnov(2010); 20472542
ModelTest,Posada和Crandall(1998); 9918953
Hyphy(v2.2),Pond等。 (2005); 15509596
Gard,Pond等。 (2006); 17110367
PAML/CODEML(v4.8),杨(2007); 17483113
红宝石(v2.3.1)
inkscape(v1.0)
PDFTEX(v3.14)
大多数Poseidon参数都是可选的,并在下面进行详细说明。
--fasta
强制的。您的输入FASTA文件必须遵循该格式:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA... >Myotis_davidii Mx1 Gene ATGGCGGTCGAGATAAGATACGTT...
所有序列都必须具有正确的开放式阅读框架,仅允许包含核苷酸字符[A | C | g | T],并且没有内部停止密码子。
序列ID必须是唯一的,直到第一次出现空间。
--reference
选修的。默认值:使用第一个序列ID作为参考。您可以从多个FASTA文件中定义一个物种ID作为参考物种。相对于该物种,将绘制积极选择的位点和相应的氨基酸。 ID必须匹配FASTA标头,直到第一个空间出现。例如,如果您希望Myotis lucifugus作为您的参考物种,而Fasta文件包含:
>Myotis_lucifugus Mx1 Gene ATGGCGATCGAGATACGATACGTA...
使用
--reference "Myotis_lucifugus"
作为设置参考物种的参数。根据默认值,将使用多个FASTA文件中的第一个ID。
--outgroup
选修的。默认:树是没有根的。您可以将一个或多个(逗号分离的)物种ID定义为外部。所有系统发育树将根据该物种植根。例如,如果您的多个Fasta文件包含
ATGGCGATCGAGATACGATACGTA...
>Myotis_davidii Mx1 Gene
ATGGCGGTCGAGATAAGATACGTT...
>Pteropus_vampyrus Mx1 Gene
ATGGCCGTAGAGATTAGATACTTT...
>Eidolon_helvum Mx1 Gene
ATGCCCGTAGAGAATAGATACTTT...您可以定义:
--outgroup "Pteropus_vampyrus,Eidolon_helvum"
与这两个物种有关的所有树木扎根。
--kh
选修的。默认值:false。使用此参数,您可以决定是否应考虑无关紧要的断点。使用Kashino木谷(KH)测试Kishino,H。和Hasegawa,M。(1989),对所有断点进行了严重的拓扑不一致测试。 KH无关紧要的断裂点最常见的是段之间的分支长度的变化。然而,考虑到KH无关紧要的断点可能很有趣,因为我们已经观察到碎片中推定的积极选择的位置而没有任何明显的拓扑不一致。 KH无关紧要的片段在最终输出中标记,因为实际重组事件可能不会发生。
根据默认,仅使用重大断点进行进一步计算。
还请记住,使用微不足道的断点也可以将波塞冬的运行时间从几分钟扩展到几小时,具体取决于检测到的断点数量。
有关其他参数(GARD,RAXML,...),请参阅--help ,并让我们知道您是否需要更多自定义!
如果波塞冬帮助您,请引用:
MartinHölzer和Manja Marz,“ Poseidon:用于检测进化重组事件和积极选择的NextFlow管道”, OUP BioInformatics (2020)


