binnacle
1.0.0
到
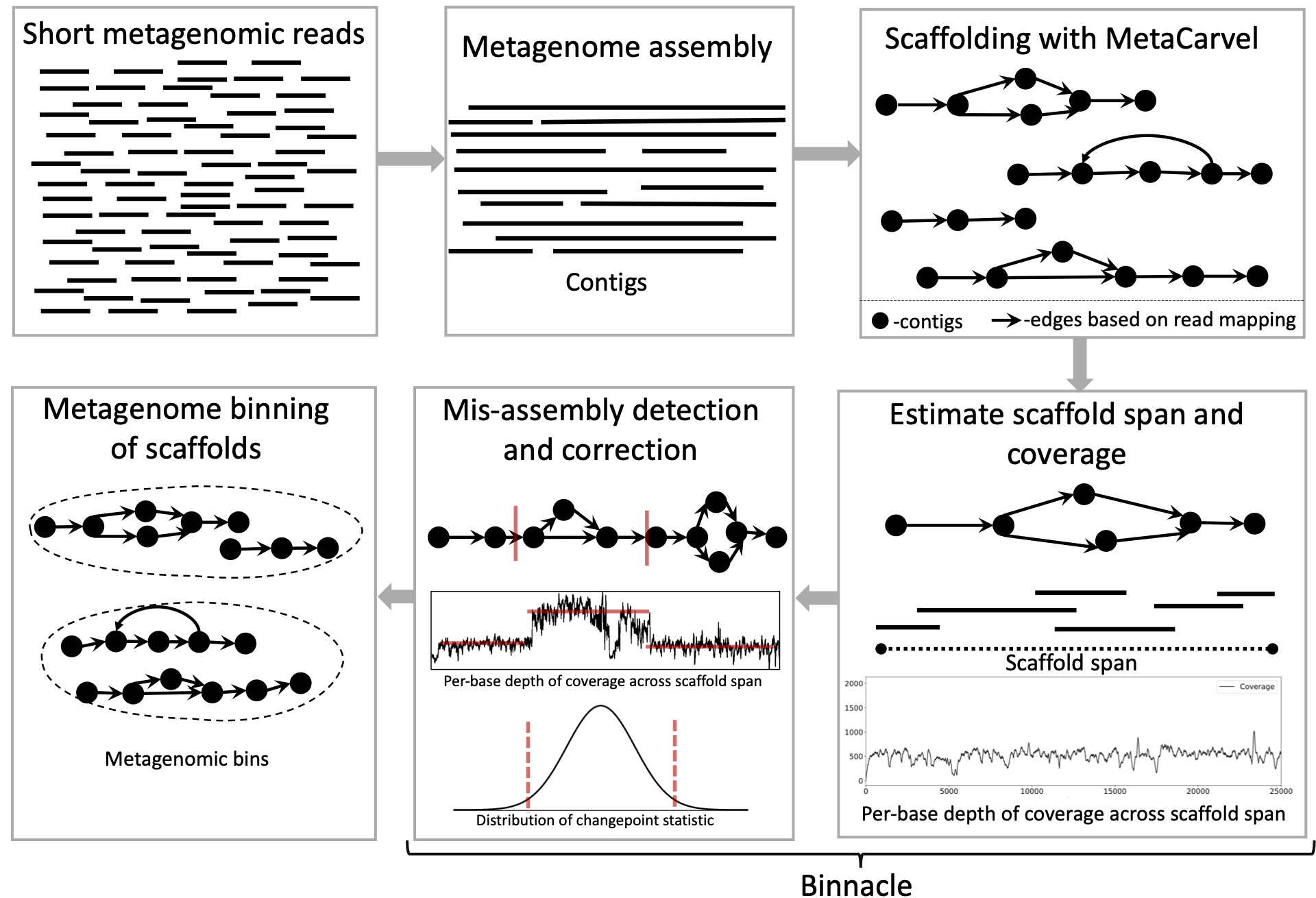
Binnacle准确地计算了图形支架的覆盖范围,并与领先的Binning方法(例如Metabat2,Maxbin 2.0和Concotct)无缝集成。使用图形支架,与重叠群(最常见的方法)相反,用于binning可以改善元基因组垃圾箱的连续性和质量,并可以捕获重建基因组的更广泛的辅助元素。
要运行Binnacle,您将需要Python 3.7.x,BedTools,Samtools,Biopython,NetworkX,Numpy和Pandas。
可以使用一个环境。这里提供了有关如何安装这些软件包的详细文档。我们使用掌脚架工具输出的图形脚手架,因此您还需要下载和安装MetAcarvel。有一个逐步安装指南,用于掌级。
通常,当您有一个或多个元基因组样品时,我们需要从每个样品中组装,脚手架和垃圾桶/支架来生成元基因组箱。我们建议将Megahit用于组装,并使用MetaCarvel进行脚手架。我们通过组装,脚手架和每位基础覆盖范围估算步骤提供了一个辅助指南。
请按照以下步骤生成文件,以便使用图形支架运行的binning方法:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
请查看Wiki,以获取有关设置Python环境的详细说明,计算覆盖范围的方法以及运行Binnacle的典型工作流程。
为了可视化图形支架,我们建议使用基于Web的浏览器Metagenomescope。 Metagenomescope的输入为assembly_graph_filtered.gml。此处给出了有关安装和运行元基因组学的详细文档。
请引用Muralidharan HS,Shah N,Meisel JS和Pop M(2021)Binnacle:使用脚手架来改善元基因组垃圾箱的连续性和质量。正面。微生物。 12:638561。 doi:10.3389/fmicb.2021.638561。
该工具仍在开发中。如果您有任何疑问,请在Github上在此处打开问题或与我们联系。
Harihara Muralidharan:[email protected]
nidhi shah:[email protected]


