ClockstaR
1.0.0

杜興斯 (Sebastian Duchene)、馬丁娜莫拉克 (Martyna Molak) 和西蒙 YW Ho。
分子生態學、演化和系統發育學 (MEEP) 實驗室
生物科學學院
雪梨大學
2015 年 6 月 10 日
partition_data_partitionfinder('drag fasta file with concatenated data here', 'drag partition finder output here')
optim.trees.interactive(folder.parts = 'path to your folder with fasta files and tree topology here')
使用 BSD 距離的導數實現樹距離的最佳化
實現拓撲距離的平行版本
編寫拓樸距離聚類教程
整合模型產生器進行模型測試
整合 RaxML 以實現分支長度和拓撲的最大似然優化
使用多基因資料集估計演化時間尺度是系統發育研究中常見的做法。多基因資料集可以按基因、密碼子位置或兩者進行分區。在本教程中,我們將「資料子集」稱為單一基因或多基因資料集的任何子單位。術語“分區”是指一組資料子集。
儘管可以使用單一鬆弛時鐘模型來連接和分析資料子集,但即使它們的樹拓撲相同,資料子集之間的譜系間速率變化模式也可能不同。例如,粒線體基因的譜系間比率變異可能與核基因的不同。因此,不同的鬆弛時鐘模型可以分配給不同的資料子集,以改進演化時間尺度和統計擬合的估計值(參見 Duchene 和 Ho.,2014a)
多基因資料集的劃分方法有很多種。比較分區方案的常見方法是使用貝葉斯因子或基於可能性的標準進行模型擬合。然而,在大多數情況下,測試所有可能的劃分方案是不可行的,尤其是使用計算貝葉斯因子的計算密集型方法。
ClockstaR 估計每個資料子集的系統發育分支長度。計算每對樹的分支分數距離(稱為 sBSDmin),作為其譜系間比率變異模式差異的測量。這些距離用於使用GAP 統計數據和PAM 聚類演算法來推斷最佳分區策略,如在包cluster 中實現的那樣(Maechler et al., 2012)(有關sBSDmin 指標的詳細信息,請參閱Duchene et al. , 2014b) 。
ClockstaR 是一個 R 軟體包,用於多基因資料集的系統發育分子時鐘分析。它使用不同基因的譜系速率變異模式來選擇時鐘分區策略。此方法使用系統發育樹距離度量和無監督機器學習演算法來確定時鐘分區的最佳數量,以及應在每個分區下分析哪些基因。 ClocsktaR 中選擇的分區策略可用於後續的分子鐘分析,例如 BEAST、MrBayes、PhyloBayes 等程式。
請點擊此連結查看原始出版物。
ClockstaR 需要安裝 R。它還需要一些 R 依賴項,這些依賴項可以透過 R 獲得,如下所述。
請將任何請求或問題發送至 Sebastian Duchene (sebastian.duchene[at]sydney.edu.au)。其他一些軟體和資源可以在雪梨大學的分子生態學、演化和系統發育實驗室找到。
下載此儲存庫作為 zip 檔案並解壓縮。以下說明使用clockstar_example_data資料夾,其中包含一些fasta檔案和newick格式的系統發育樹。在文字編輯器(例如 text wrangler)中開啟這些文件中的任何一個。這些數據是在四種進化速率變化模式下模擬的。請注意,樹是所有基因或資料分區的樹拓撲。要執行 ClockstaR,請格式化您的數據,類似於 Clockstar_example_data 中的範例資料。
ClockstaR 可以直接從 GitHub 安裝。這需要 devtools 套件。在 R 提示字元處鍵入以下程式碼以安裝所有必要的工具(請注意,您需要網路連線才能直接下載軟體套件):。
install . packages ( " devtools " )
library (devtools)
install_github ( ' ClockstaR ' , ' sebastianduchene ' )下載並安裝後,載入ClockstaR的函式庫。
library (ClockstaR2)若要查看程式如何運作的範例,請輸入:
example (ClockstaR2)本教學的其餘部分使用 Clockstar_example_data 資料夾
第一步是取得每個比對的基因樹。為此,我們使用樹拓撲,並使用每個單獨的基因比對(在本例中為 A1.fasta 到 C3.fasta)來最佳化分支長度。如果您有基因樹,則以 newick 格式儲存在檔案中,然後進入下一步(互動式執行 Clockstar)。
在 R 提示符號中輸入以下代碼並按 Enter 鍵:
optim . trees . interactive ()如果您收到有關安裝 phangorn 套件的錯誤訊息,請使用此程式碼,然後重複 optim.trees.interactive()
install . packcages ( " phangorn " )ClockstaR 將列印以下訊息:
Please drag a folder with the data subsets and a tree topology . The files should be in FASTA format, and the trees in NEWICK將 Clockstar_example_data 資料夾拖曳到 R 控制台並輸入 Enter。請注意,該資料夾應僅包含 FASTA 格式的排列和 NEWICK 格式的樹狀拓撲。您將看到以下訊息:
What should be the name of the file to save the optimised trees ?輸入優化樹的檔案名稱。在這種情況下,我們將使用“example.trees”
example . trees此時,ClockstaR 會詢問是否應該對每個基因使用單獨的替換模型,或在所有情況下都使用 JC。由於這些數據是在 JC 下模擬的,因此我們將鍵入“n”並按 Enter 鍵。鍵入“y”以分別指定每個替代模型。
輸入「n」並按 Enter 鍵後,ClockstaR 將開始運作。它將在圖形設備中列印基因樹。如果指定的樹已紮根,它還可能會列印一些警告,可以安全地忽略這些警告。
打開clockstar_example_data 資料夾。您將找到一個名為“example.trees”的文件,如上幾個步驟所指定。在文字編輯器中開啟 example.trees。它包含每個基因樹和根據基因比對名稱的樹名稱。它應該看起來像這樣:
A1 . fasta (( t1 : 0.01504695462 ,( t2 : 0.00987 ...
A2 . fasta (( t1 : 0.01520523401 ,( t2 : 0.01317 ...
A3 . fasta (( t1 : 0.01519309467 ,( t2 : 0.01092 ...
.
.
.該包含樹的檔案將用於下一步。
對於此步驟,必須將基因樹保存在檔案中,例如在上一個步驟中獲得的檔案。
打開 R 並載入 ClockstaR,如上所示。在提示符號下鍵入以下程式碼:
clockstar . interactive ()ClockstaR 將列印以下訊息:
please drag or type in the path to your gene trees file in NEWICK format :將帶有基因樹的檔案拖曳到 R 控制台。如果您按照上一個步驟操作,該檔案將被稱為 example.trees。輸入回車。
根據您安裝的軟體包,ClockstaR 可能會詢問是否應該並行運行。這對於大型資料集非常有效。但對於範例資料來說,它不會產生很大的差異,因此如果您看到此訊息,請輸入“n”,然後輸入 Enter:
Packages foreach and doParallel are available for parallel computation
Should we run ClockstaR in parallel (y / n) ? (This is good for large data sets)Clockstar 現在將開始運作。螢幕上的輸出應如下所示:
[ 1 ] " Calculating sBSDmin distances between all pairs of trees "
[ 1 ] " Estimating tree distances "
[ 1 ] " estimating distances 1 of 11 "
[ 1 ] " estimating distances 2 of 11 "
[ 1 ] " estimating distances 3 of 11 "
[ 1 ] " estimating distances 4 of 11 "
[ 1 ] " estimating distances 5 of 11 "
.
.
.估計樹距離(在原始出版物中描述)後,ClockstaR 將列印以下訊息:
" I finished calculating the sBSDmin distances between trees "
The settings for clustering with ClockstaR are :
PAM clustering algorithm
K from 1 to number of data subsets - 1
SEmax criterion to select the optimal k
500 bootstrap replicates
Are these correct ? (y / n)這些是聚類演算法的設定。它們適用於大多數資料集,因此在本例中我們可以鍵入“y”,然後輸入。
ClockstaR 現在將運行聚類演算法。最後它會列印最佳分區數並詢問是否應將結果保存在 pdf 文件中:
[ 1 ] " ClockstaR has finished running "
[ 1 ] " The best number of partitions for your data set is: 3 "
Do you wish to save the results in a pdf file ? (y / n)輸入“y”然後回車。
然後 ClockstaR 將詢問輸出檔案的名稱:
What should be the name and path of the output file ?對於此範例,鍵入“example_run”並回車,但可以使用任何名稱。
現在開啟clockstar_example_data資料夾並開啟兩個pdf檔example_run_gapstats.pdf和example_run_matrix.pdf。
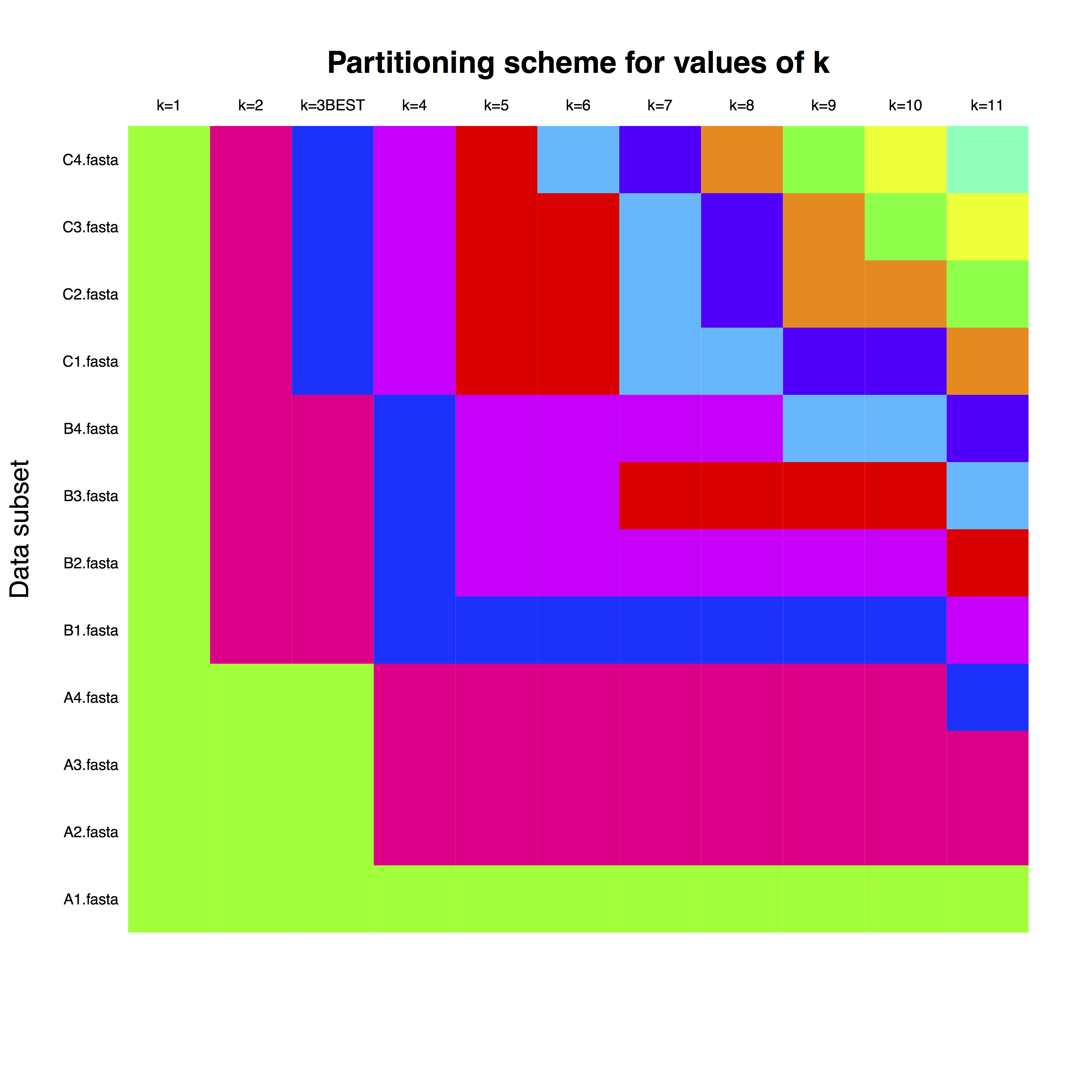
example_run_matrix 是矩陣,其中行對應於每個基因,如 FASTA 檔案中所命名。列是分區的數量,顏色代表每個基因對時鐘分區的分配。例如,對於k = 3(這是最佳分區數),可以對具有字母 A、B 和 C 的基因使用單獨的時鐘分區。
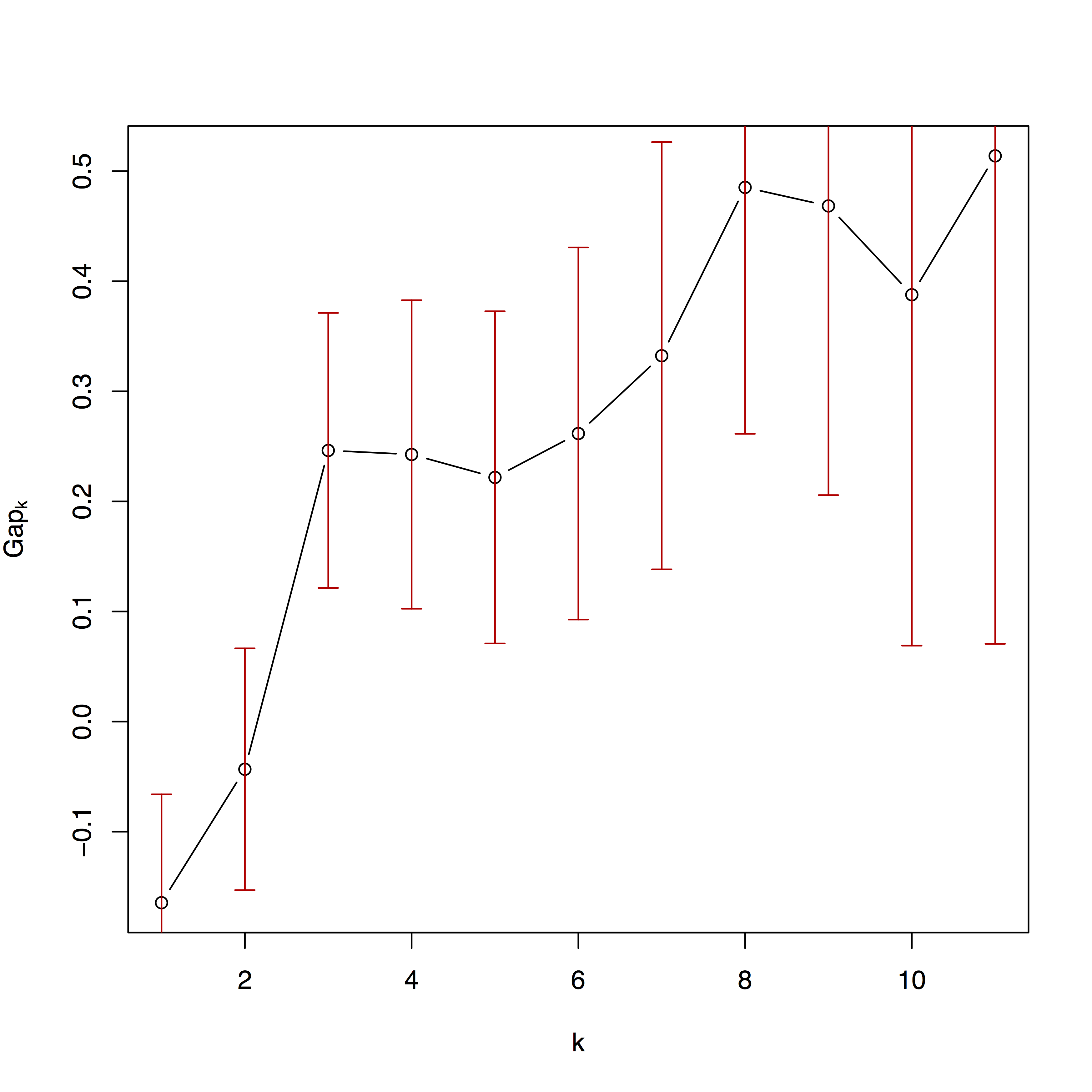
第二張圖是聚類演算法在不同數量的分區上的擬合情況。更多詳細資訊請參閱 Kaufman 和 Rousseeuw (2009) 以及包 cluster 的文檔。
ClockstaR 可以使用其他自訂設定來運作。請參閱文件以了解其他詳細信息,如果有任何問題,請致電 sebastian.duchene[at]sydney.edy.au。
標誌由童俊設計
Duchene, S. 與 Ho, SY (2014a)。使用多個鬆弛時鐘模型根據 DNA 序列資料估計進化時間尺度。分子系統發育與演化(77):65-70。
Duchene, S.、Molak, M. 與 Ho, SY (2014b)。 ClockstaR:在分子系統發育分析中選擇鬆弛時鐘模型的數量。生物資訊學30 (7): 1017-1019。
考夫曼,L.,和盧梭,PJ (2009)。尋找資料中的群組:聚類分析簡介(第 344 卷)。約翰威利父子。


