VARUS
1.0.0
Varus最初是由威利·布魯恩(Willy Bruhn)撰寫的,是馬里奧·斯坦克(Mario Stanke)監督的學士學位論文。該存儲庫是https://github.com/willybruhn/varus於2018年11月製作的副本,其中包含許多錯誤文件,增量內含子數據庫功能以及用於使用HISAT AL替代對準程序的擴展程序。
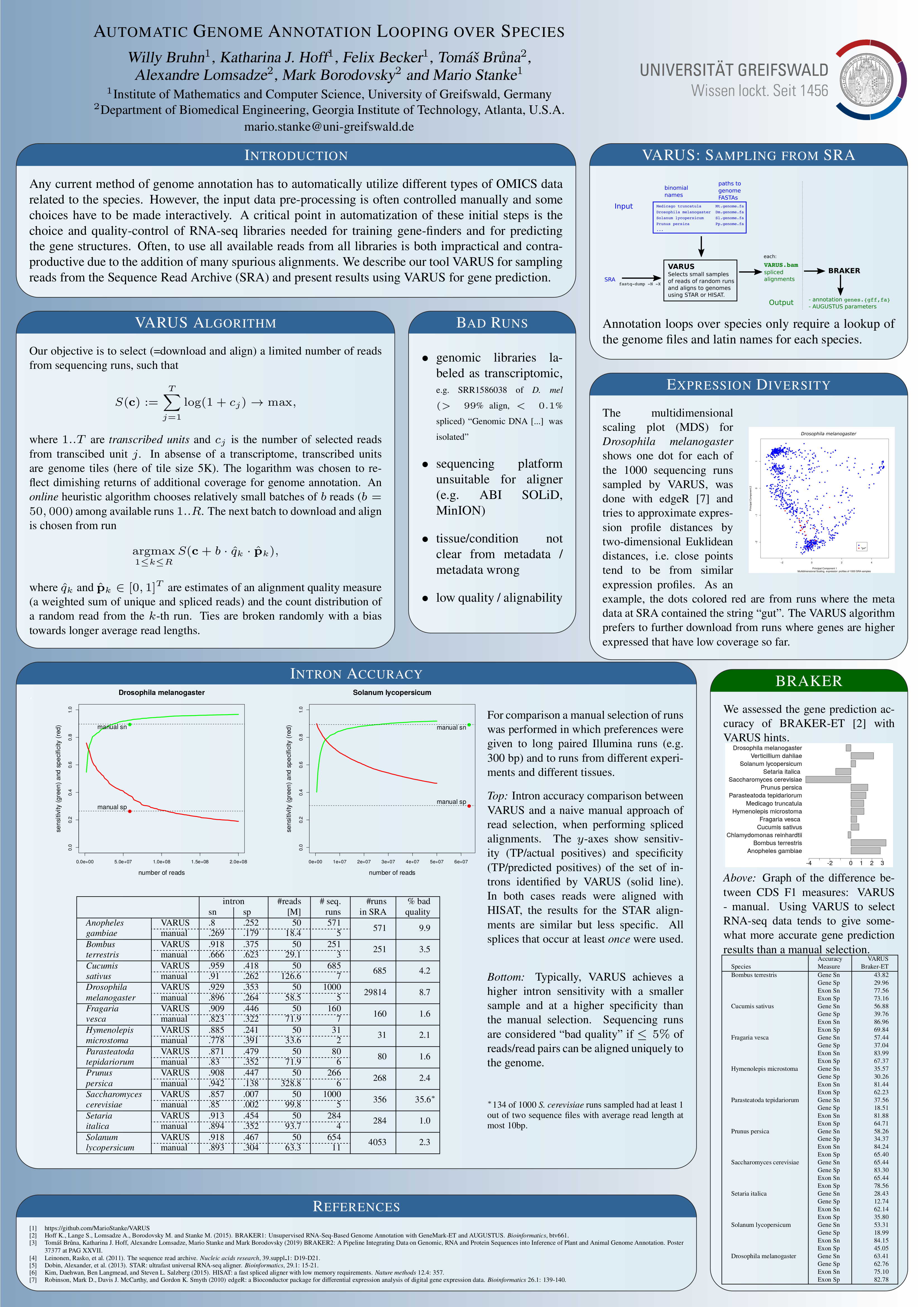
VARUS自動選擇和下載有限數量的RNA-seq讀取的NCBI序列讀取檔案(SRA),其針對許多基因的覆蓋範圍足夠高,以進行基因 - 芬德訓練和基因組註釋。在線算法的每次迭代
調用命令行中的以下命令以克隆存儲庫:
git clone https://github.com/MarioStanke/VARUS.git變量取決於
sudo apt-get install bamtools libbamtools-dev手動編譯雜色
cd Implementation
make
默認情況下,NCBI工具fastq-dump在~/ncbi下創建與下載數據的運行文件相同的臨時文件,即使僅下載了一個小部分。禁用這種緩存行為,對於大多數使用
mkdir -p ~/.ncbi
echo '/repository/user/cache-disabled = "true"' >> ~/.ncbi/user-settings.mkfg
更改為目錄example ,並按照示例/讀書中的說明進行操作。
將文件VARUSparameters.txt從示例文件夾複製到您的工作目錄,並在必要時進行調整:
最重要的參數:
- 截面規定在每次迭代中應下載多少次讀取(例如50000或200000)
- 最大鍵盤指定最多應下載多少批次
最終輸出是一個分類的剪接對齊文件(所有批次)稱為varus.bam 。
請引用:VARUS:採樣互補的RNA從序列讀取存檔中讀取。 2019; BMC生物信息學,20:558
找到對應於 /doc /論文的變量的威利·布魯恩(Willy Bruhn)的學士學位。


