Dieses Repo enthält:
sepal erfordert python3 , vorzugsweise eine Version höher oder gleich 3.5. Öffnen Sie zum Herunterladen und Installieren das Terminal, wechseln Sie in das Verzeichnis, in das sepal heruntergeladen werden soll, und gehen Sie wie folgt vor:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
Abhängig von Ihren Benutzerrechten müssen Sie möglicherweise --user als Argument zu setup.py hinzufügen. Wenn Sie das Setup ausführen, erhalten Sie die minimal erforderliche Installation, um die Diffusionszeiten zu berechnen. Wenn Sie jedoch die Analysemodule nutzen möchten, müssen Sie zusätzlich die empfohlenen Pakete installieren. Führen Sie dazu einfach (im selben Verzeichnis) Folgendes aus:
pip install -e " .[full] " Auch hier könnte es notwendig sein, den --user einzuschließen. Außerdem müssen Sie möglicherweise pip3 verwenden, wenn Sie Ihre python-pip -Schnittstelle auf diese Weise eingerichtet haben. Wenn Sie conda oder virtuelle Umgebungen verwenden, befolgen Sie deren Empfehlungen zur Installation von Paketen.
Dadurch sollten sowohl eine Befehlszeilenschnittstelle (CLI) als auch ein Standardpaket installiert werden. Um zu testen, ob die Installation erfolgreich war, können Sie versuchen, den folgenden Befehl auszuführen:
sepal -h
Dadurch sollte die dem Kelchblatt zugeordnete Hilfemeldung ausgegeben werden. Wenn bisher alles für Sie geklappt hat, können Sie mit dem Beispielabschnitt fortfahren, um sepal in Aktion zu sehen!
Die empfohlene Verwendung von Sepal erfolgt über die Befehlszeilenschnittstelle. Sowohl die Simulationen zur Berechnung der Diffusionszeiten als auch die anschließende Analyse oder Überprüfung der Ergebnisse können einfach durchgeführt werden, indem man sepal gefolgt von run oder analyze eingibt. Das analyze verfügt über verschiedene Optionen, um die Ergebnisse zu visualisieren ( inspect ), die Profile in Musterfamilien zu sortieren ( family ) oder die identifizierten Familien einer funktionalen Anreicherungsanalyse zu unterziehen ( fea ). Um eine vollständige Liste der verfügbaren Befehle zu erhalten, führen Sie sepal module -h aus, wobei module entweder run oder analyze ist. Im Folgenden veranschaulichen wir, wie Kelchblätter verwendet werden können, um Transkriptionsprofile mit räumlichen Mustern zu finden.
Wir erstellen einen Ordner zur Speicherung unserer Ergebnisse, der auch als unser Arbeitsverzeichnis fungiert. Gehen Sie im Hauptverzeichnis des Repos wie folgt vor:
cd res
mkdir example
cd exampleDie MOB-Probe wird zur Veranschaulichung unserer Analyse verwendet. Wir beginnen mit der Berechnung der Diffusionszeiten für jedes Transkriptionsprofil:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1Unten sehen Sie ein Beispiel (mit zusätzlicher Anzeige des Hilfebefehls), wie dies aussehen könnte

Nachdem wir die Diffusionszeiten berechnet haben, wollen wir uns das Ergebnis ansehen, indem wir uns wie in der Studie die Top-20-Profile ansehen. Wir können ganz einfach Bilder aus unserem Ergebnis generieren, indem wir den folgenden Befehl ausführen:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5Das würde in etwa so aussehen:

Die Ausgabe wird das folgende Bild sein:
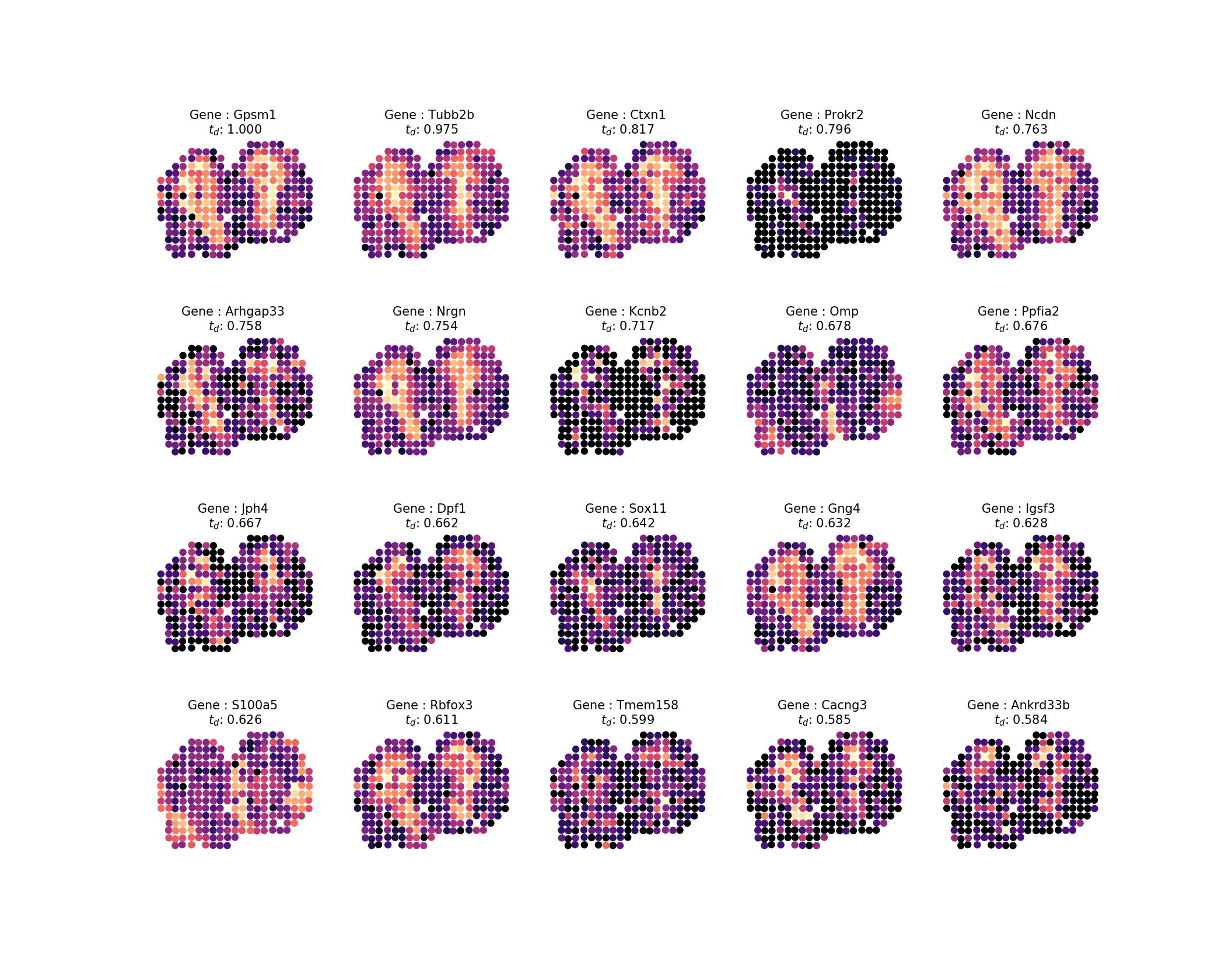
Um dann die 100 am höchsten bewerteten Gene in eine Reihe von Musterfamilien zu sortieren, wobei 85 % der Varianz in unseren Mustern durch die Eigenmuster erklärt werden sollten, gehen Sie wie folgt vor:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
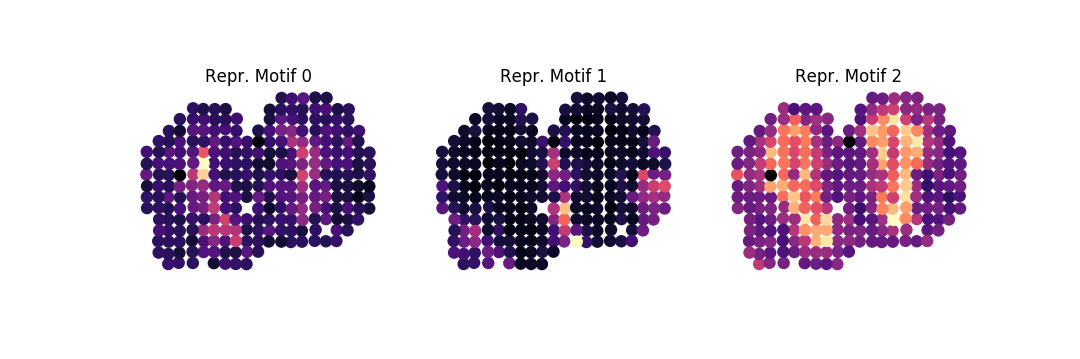
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3Daraus ergeben sich für jede Familie folgende drei repräsentative Motive:
Wir können unsere Familien einer Bereicherungsanalyse unterziehen, indem wir Folgendes durchführen:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "wo wir zum Beispiel sehen, dass Familie 2 für mehrere Prozesse im Zusammenhang mit neuronaler Funktion, Erzeugung und Regulierung bereichert ist:
| Familie | einheimisch | Name | p_value | Quelle | Kreuzungsgröße | |
|---|---|---|---|---|---|---|
| 2 | 2 | GO:0007399 | Entwicklung des Nervensystems | 0,00035977 | GO:BP | 26 |
| 3 | 2 | GO:0050773 | Regulierung der Dendritenentwicklung | 0,000835883 | GO:BP | 8 |
| 4 | 2 | GO:0048167 | Regulierung der synaptischen Plastizität | 0,00196494 | GO:BP | 8 |
| 5 | 2 | GO:0016358 | Dendritenentwicklung | 0,00217167 | GO:BP | 9 |
| 6 | 2 | GO:0048813 | Dendritenmorphogenese | 0,00741589 | GO:BP | 7 |
| 7 | 2 | GO:0048814 | Regulierung der Dendritenmorphogenese | 0,00800399 | GO:BP | 6 |
| 8 | 2 | GO:0048666 | Neuronenentwicklung | 0,0114088 | GO:BP | 16 |
| 9 | 2 | GO:0099004 | Calmodulin-abhängiger Kinase-Signalweg | 0,0159572 | GO:BP | 3 |
| 10 | 2 | GO:0050804 | Modulation der chemischen synaptischen Übertragung | 0,0341913 | GO:BP | 10 |
| 11 | 2 | GO:0099177 | Regulierung der transsynaptischen Signalübertragung | 0,0347783 | GO:BP | 10 |
Selbstverständlich erhebt diese Analyse keinen Anspruch auf Vollständigkeit. Vielmehr ein kurzes Beispiel, um zu zeigen, wie man die CLI für sepal bedient.
Obwohl sepal als eigenständiges Tool konzipiert wurde, haben wir es auch so konzipiert, dass es als Standard-Python-Paket funktioniert, aus dem Funktionen importiert und in einem integrierten Workflow verwendet werden können. Um zu zeigen, wie dies geschehen kann, geben wir ein Beispiel und reproduzieren die Melanomanalyse. Weitere Beispiele können später hinzugefügt werden.
Die Eingabe in sepal muss im Format n_locations x n_genes erfolgen. Wenn Ihre Daten jedoch umgekehrt strukturiert sind ( n_genes x n_locations ), geben Sie einfach das Flag --transpose an, wenn Sie die Simulation oder Analyse ausführen. Dies wird dann berücksichtigt von.
Wir unterstützen derzeit die Formate .csv , .tsv und .h5ad . Für Letzteres sollte Ihre Datei nach DIESEM Format strukturiert sein. Wir gehen davon aus, dass das scanpy -Team in naher Zukunft eine Veröffentlichung veröffentlichen wird, in der ein standardisiertes Format für Geodaten vorgestellt wird. Bis dahin werden wir jedoch den oben genannten Standard verwenden.
Alle von uns verwendeten realen Daten sind öffentlich und können unter den folgenden Links abgerufen werden:
Die synthetischen Daten wurden generiert von:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py Alle in der Studie präsentierten Ergebnisse sind im res -Ordner zu finden, sowohl für die realen als auch für die synthetischen Daten. Für jede Probe haben wir die Ergebnisse entsprechend strukturiert:
res/sample-name/X-diffusion-times.tsv : Diffusionszeiten für alle rangierten Geneanalysis/ : enthält die Ausgabe der Sekundäranalyse

