binnacle
1.0.0
Ke
Binnacle secara akurat menghitung cakupan perancah grafik dan diintegrasikan dengan mulus dengan metode binning terkemuka seperti Metabat2, Maxbin 2.0, dan CHEAT. Menggunakan perancah grafik, sebagai lawan dari contigs (pendekatan paling umum) untuk binning meningkatkan kedekatan dan kualitas tempat sampah metagenomik dan dapat menangkap serangkaian elemen aksesori yang lebih luas dari genom yang direkonstruksi.
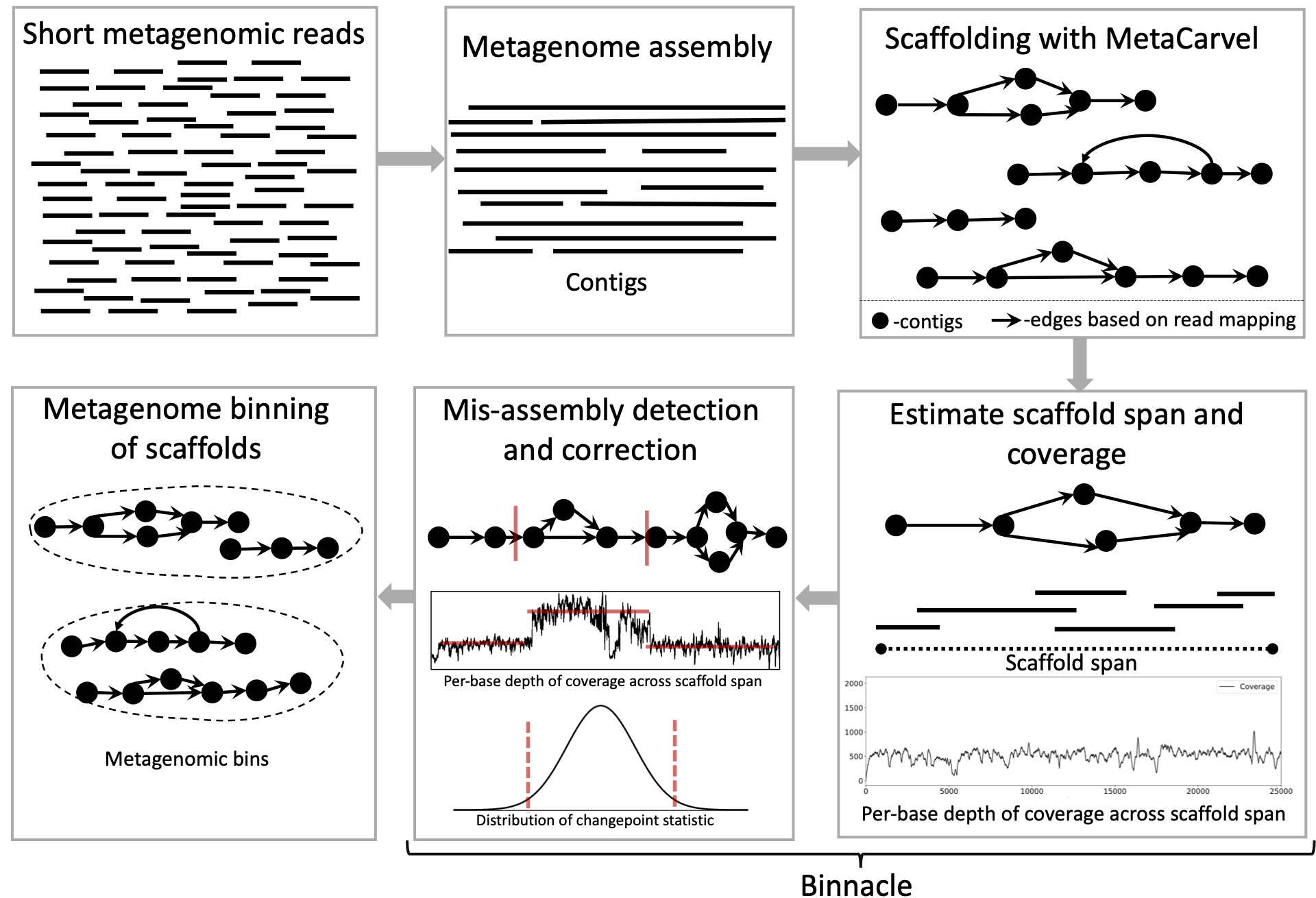
Untuk menjalankan binnacle, Anda akan membutuhkan Python 3.7.x, bedtools, samtools, biopython, networkx, numpy, dan panda.
File lingkungan.yml tersedia dan ini dapat digunakan untuk menciptakan lingkungan Conda yang cocok untuk menjalankan binnacle. Dokumentasi terperinci tentang cara menginstal paket -paket ini diberikan di sini. Kami menggunakan perancah grafik yang merupakan output dari alat perancah Metacarvel, jadi Anda juga perlu mengunduh dan menginstal MetaCarvel. Ada panduan instalasi langkah demi langkah untuk Metacarvel.
Secara umum, ketika Anda memiliki satu atau beberapa sampel metagenomik, kita perlu berkumpul, perancah, dan nampan/perancah dari setiap sampel untuk menghasilkan tempat sampah metagenomik. Kami merekomendasikan menggunakan Megahit untuk perakitan, dan Metacarvel untuk perancah. Kami menyediakan panduan pembantu untuk bekerja melalui perakitan, perancah, dan langkah-langkah estimasi pertanggungan per-base di sini.
Ikuti langkah -langkah ini untuk menghasilkan file untuk menjalankan metode binning dengan perancah grafik:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
Silakan periksa wiki untuk deskripsi terperinci tentang pengaturan lingkungan Python, metode untuk menghitung cakupan dan alur kerja khas untuk menjalankan binnacle.
Untuk memvisualisasikan perancah grafik, kami sarankan menggunakan Metagenomescope yang merupakan browser berbasis web. Input ke Metagenomescope adalah Assembly_Graph_filtered.gml. Dokumentasi terperinci tentang menginstal dan menjalankan metagenomescope diberikan di sini.
Tolong kutip Muralidharan HS, Shah N, Meisel JS dan Pop M (2021) Binnacle: Menggunakan perancah untuk meningkatkan kedekatan dan kualitas tempat sampah metagenomik. Depan. Mikrobiol. 12: 638561. doi: 10.3389/fmicb.2021.638561.
Alat ini masih sedang dikembangkan. Harap buka masalah di sini di GitHub atau hubungi kami jika Anda memiliki pertanyaan.
Harihara Muralidharan: [email protected]
Nidhi Shah: [email protected]


