methylbert
v.2.0.1_doi
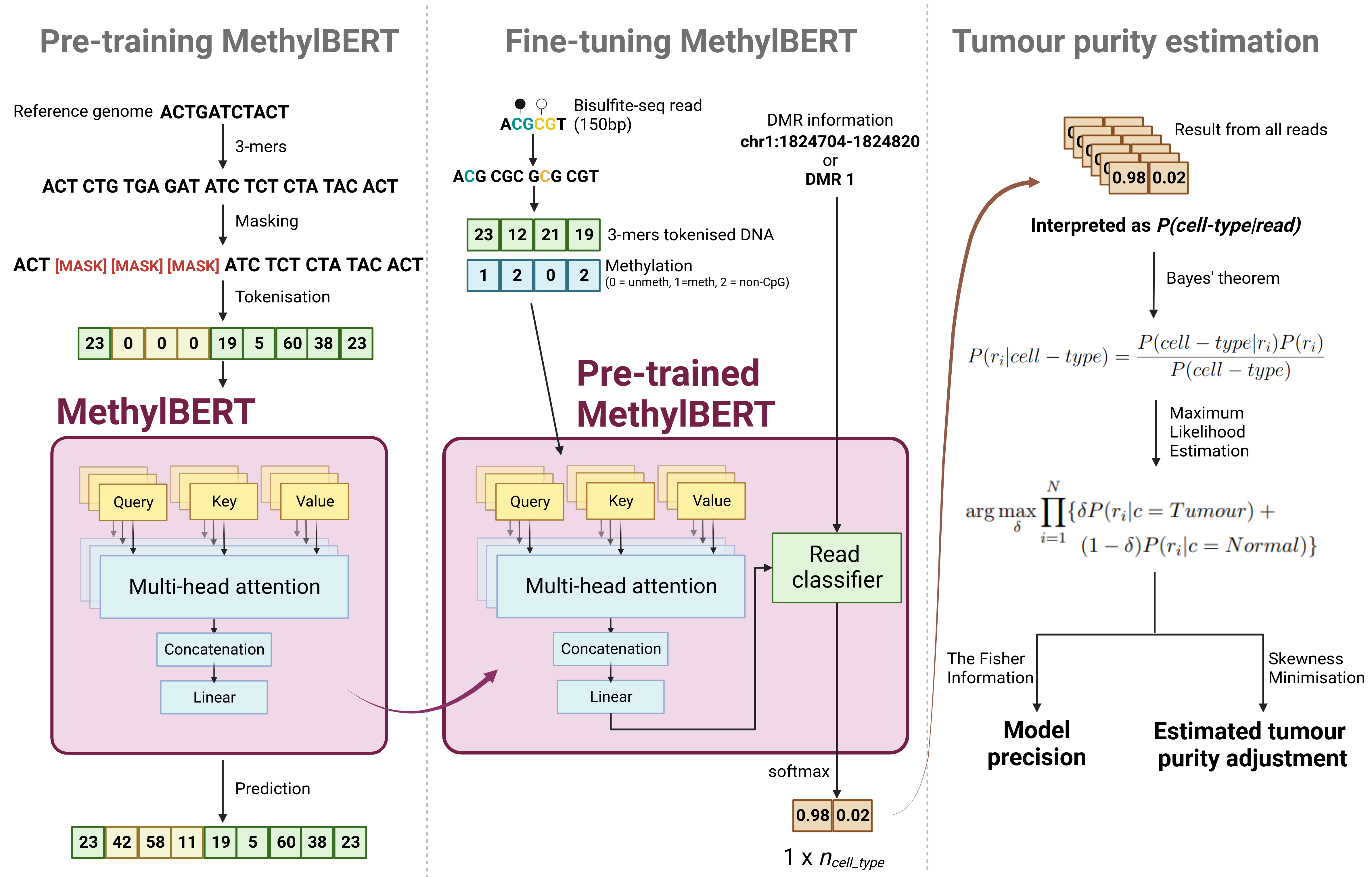 methylbert _scheme" style="max-width: 100%;"> ตัวเลขถูกสร้างขึ้นโดยใช้ biorender
methylbert _scheme" style="max-width: 100%;"> ตัวเลขถูกสร้างขึ้นโดยใช้ biorender
แบบจำลอง BERT เพื่อจัดประเภทข้อมูลเมทิลเลชันของ DNA ระดับที่อ่านได้เป็นเนื้องอก/ปกติ และดำเนินการแยกส่วนเนื้องอก methylbert ถูกนำไปใช้โดยใช้ pytorch และหม้อแปลงไฟฟ้า ?
methylbert paper ออนไลน์แล้วบน bioRxiv !!
methylbert : แบบจำลองที่ใช้หม้อแปลงไฟฟ้าสำหรับการระบุรูปแบบเมทิลเลชั่นของ DNA ระดับการอ่านและการสลายตัวของเนื้องอก, Yunhee Jeong, Karl Rohr, Pavlo Lutsik, bioRxiv 2023.10.29.564590; ดอย : https://doi.org/10.1101/2023.10.29.564590
methylbert ทำงานได้อย่างเสถียรที่สุดด้วย Python=3.11
methylbert มีให้เป็นแพ็คเกจหลาม
conda create -n methylbert -c conda-forge python=3.11 cudatoolkit==11.8 pip freetype-py
conda activate methylbert
pip install methylbert
คุณสามารถตั้งค่าสภาพแวดล้อม conda ของคุณด้วยไฟล์ environment.yml ได้โดยการรัน conda env create --file environment.yml หรือแทน:
conda create -n methylbert -c conda-forge python=3.11 cudatoolkit==11.8 pip freetype-py
conda activate methylbert
git clone https://github.com/hanyangii/methylbert.git
cd methylbert
pip3 install .
หากคุณต้องการใช้ methylbert เป็นไลบรารี่ของ Python โปรดปฏิบัติตามบทช่วยสอน
methylbert รองรับเครื่องมือบรรทัดคำสั่ง ก่อนที่จะใช้เครื่องมือบรรทัดคำสั่ง โปรดตรวจสอบข้อกำหนดของไฟล์อินพุต
> methylbert
methylbert v2.0.1
One option must be given from ['preprocess_finetune', 'finetune', 'deconvolute']
-h หรือ --help ให้อาร์กิวเมนต์ที่มีอยู่สำหรับแต่ละฟังก์ชัน (เช่น methylbert preprocess_finetune --help )
เช่น) methylbert preprocess_finetune -f bulk.bam -d dmrs.csv -r genome.fa -p 0.8 -c 50 -o data/
-s SC_DATASET, --sc_dataset SC_DATASET
a file all single-cell bam files are listed up. The first and second columns must indicate file names and cell types if cell types are given. Otherwise, each line must have one file path.
-f INPUT_FILE, --input_file INPUT_FILE
.bam file to be processed
-d F_DMR, --f_dmr F_DMR
.bed or .csv file DMRs information is contained
-o OUTPUT_PATH, --output_path OUTPUT_PATH
a directory where all generated results will be saved
-r F_REF, --f_ref F_REF
.fasta file containing reference genome
-nm N_MERS, --n_mers N_MERS
K for K-mer sequences (default: 3)
-m METHYLCALLER, --methylcaller METHYLCALLER
Used methylation caller. It must be either bismark or dorado (default: bismark)
-p SPLIT_RATIO, --split_ratio SPLIT_RATIO
the ratio between train and test dataset (default: 0.8)
-nd N_DMRS, --n_dmrs N_DMRS
Number of DMRs to take from the dmr file. If the value is not given, all DMRs will be used
-c N_CORES, --n_cores N_CORES
number of cores for the multiprocessing (default: 1)
--seed SEED random seed number (default: 950410)
--ignore_sex_chromo IGNORE_SEX_CHROMO
Whether DMRs at sex chromosomes (chrX and chrY) will be ignored (default: True)
เช่น) methylbert finetune -c data/train_seq.csv -t data/test_seq.csv -o model/ -l 12 -s 150 -b 256 --gradient_accumulation_steps 4 -e 600 -w 8 --log_freq 1 --eval_freq 1 --warm_up 1 --lr 1e-4 --decrease_steps 200
-c TRAIN_DATASET, --train_dataset TRAIN_DATASET
train dataset for train bert
-t TEST_DATASET, --test_dataset TEST_DATASET
test set for evaluate train set
-o OUTPUT_PATH, --output_path OUTPUT_PATH
ex)output/bert.model
-p PRETRAIN, --pretrain PRETRAIN
path to the saved pretrained model to restore
-l N_ENCODER, --n_encoder N_ENCODER
number of encoder blocks. One of [12, 8, 6] need to be given. A pre-trained methylbert model is downloaded accordingly. Ignored when -p (--pretrain) is given.
-nm N_MERS, --n_mers N_MERS
n-mers (default: 3)
-s SEQ_LEN, --seq_len SEQ_LEN
maximum sequence len (default: 150)
-b BATCH_SIZE, --batch_size BATCH_SIZE
number of batch_size (default: 50)
--valid_batch VALID_BATCH
number of batch_size in valid set. If it's not given, valid_set batch size is set same as the train_set batch size
--corpus_lines CORPUS_LINES
total number of lines in corpus
--loss LOSS Loss function for fine-tuning. It can be either 'bce' or 'focal_bce' (default: bce)
--max_grad_norm MAX_GRAD_NORM
Max gradient norm (default: 1.0)
--gradient_accumulation_steps GRADIENT_ACCUMULATION_STEPS
Number of updates steps to accumulate before performing a backward/update pass. (default: 1)
-e STEPS, --steps STEPS
number of training steps (default: 600)
--save_freq SAVE_FREQ
Steps to save the interim model
-w NUM_WORKERS, --num_workers NUM_WORKERS
dataloader worker size (default: 20)
--with_cuda WITH_CUDA
training with CUDA: true, or false (default: True)
--log_freq LOG_FREQ Frequency (steps) to print the loss values (default: 100)
--eval_freq EVAL_FREQ
Evaluate the model every n iter (default: 10)
--lr LR learning rate of adamW (default: 4e-4)
--adam_weight_decay ADAM_WEIGHT_DECAY
weight_decay of adamW (default: 0.01)
--adam_beta1 ADAM_BETA1
adamW first beta value (default: 0.9)
--adam_beta2 ADAM_BETA2
adamW second beta value (default: 0.98)
--warm_up WARM_UP steps for warm-up (default: 100)
--decrease_steps DECREASE_STEPS
step to decrease the learning rate (default: 200)
--seed SEED seed number (default: 950410)
เช่น) methylbert deconvolute -i data/data.txt -m model/ -o res/ -b 128 --adjustment
-i INPUT_DATA, --input_data INPUT_DATA
Bulk data to deconvolute
-m MODEL_DIR, --model_dir MODEL_DIR
Trained methylbert model
-o OUTPUT_PATH, --output_path OUTPUT_PATH
Directory to save deconvolution results. (default: ./)
-b BATCH_SIZE, --batch_size BATCH_SIZE
Batch size. Please decrease the number if you do not have enough memory to run the software (default: 64)
--save_logit Save logits from the model (default: False)
--adjustment Adjust the estimated tumour purity (default: False)
@article {Jeong2023methyl,
author = {Jeong, Yunhee and Gerh{"a}user, Clarissa and Sauter, Guido and Schlomm, Thorsten and Rohr, Karl and Lutsik, Pavlo},
title = { methylbert : A Transformer-based model for read-level DNA methylation pattern identification and tumour deconvolution},
elocation-id = {2023.10.29.564590},
year = {2024},
doi = {10.1101/2023.10.29.564590},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2024/08/29/2023.10.29.564590},
eprint = {https://www.biorxiv.org/content/early/2024/08/29/2023.10.29.564590.full.pdf},
journal = {bioRxiv}
}


