BRAKER
v3.0.8
นี่คือการบันทึกการประชุมเชิงปฏิบัติการ BGA23 ครั้งแรกใน Braker หากการเรียนรู้โดยการดูวิดีโอเป็นเรื่องง่ายสำหรับคุณลองดูว่า: https://www.youtube.com/watch?v=uxtkj4mukyg
Braker3 อยู่ใน https://usegalaxy.eu/
TSEBRA & BRAKER3 ที่เกี่ยวข้อง:
Braker & Augustus ที่เกี่ยวข้อง:
Genemark เกี่ยวข้อง:
Mark Borodovsky, Georgia Tech, USA, [email protected]
Tomas Bruna สถาบันจีโนมร่วม, สหรัฐอเมริกา, [email protected]
Alexandre Lomsazde, Georgia Tech, USA, [email protected]
[A] University of Greifswald, สถาบันคณิตศาสตร์และวิทยาศาสตร์คอมพิวเตอร์, Walther-Rathenau-STR 47, 17489 Greifswald ประเทศเยอรมนี
[B] มหาวิทยาลัย Greifswald, ศูนย์จีโนมการทำงานของจุลินทรีย์, Felix-Hausdorff-Str 8, 17489 Greifswald, เยอรมนี
[C] ร่วม Georgia Tech และ Emory University Wallace H Coulter ภาควิชาวิศวกรรมชีวการแพทย์, 30332 แอตแลนตา, สหรัฐอเมริกา
[D] School of Computational Science and Engineering, 30332 Atlanta, USA
[E] สถาบันฟิสิกส์และเทคโนโลยีมอสโก, มอสโกภูมิภาค 141701, Dolgoprudny, รัสเซีย
![Braker2-Team-2 [Fig10]](https://images.downcodes.com/uploads/20250214/img_67aee79a0cb7530.png)
![Braker2-Team-1 [Fig11]](https://images.downcodes.com/uploads/20250214/img_67aee79a0d1eb31.png)
![Braker2-Team-3 [รูปที่ 12]](https://images.downcodes.com/uploads/20250214/img_67aee79a0da9c32.png)
![Braker2-Team-4 [รูปที่ 13]](https://images.downcodes.com/uploads/20250214/img_67aee79a0e49933.png)
รูปที่ 1: ผู้เขียนเบเกอร์ปัจจุบันจากซ้ายไปขวา: Mario Stanke, Alexandre Lomsadze, Katharina J. Hoff, Tomas Bruna, Lars Gabriel และ Mark Borodovsky เรารับทราบว่าชุมชนนักวิทยาศาสตร์ขนาดใหญ่มีส่วนร่วมในรหัส Braker (เช่นผ่านคำขอดึง)
การพัฒนาของ Braker1, Braker2 และ Braker3 ได้รับการสนับสนุนจากสถาบันสุขภาพแห่งชาติ (NIH) [GM128145 ถึง MB และ MS] การพัฒนาของ Braker3 ได้รับการสนับสนุนบางส่วนจากความสามารถของข้อมูลโครงการที่มอบให้แก่ KJH และ MS โดยรัฐบาลของ Mecklenburg-Vorpommern ประเทศเยอรมนี
ตัวเลือกการถอดเสียงสำหรับ Braker (TSEBRA) มีอยู่ที่ https://github.com/gaius-augustus/tsebra
Genemark-ETP หนึ่งในตัวค้นหายีนที่เป็นแกนหลักของ Braker มีให้บริการที่ https://github.com/gatech-genemark/genemark-etp
ออกัสตัสตัวค้นหายีนที่สองที่เป็นแกนหลักของเบเกอร์มีให้บริการที่ https://github.com/gaius-augustus/augustus
Galba ซึ่งเป็นเครื่องปั่นปัดเบเกอร์สำหรับใช้ miniprot หรือ genomethreader เพื่อสร้างยีนการฝึกอบรมมีให้ที่ https://github.com/gaius-augustus/galba
จำนวนจีโนมที่มีการจัดลำดับที่เพิ่มขึ้นอย่างรวดเร็วนั้นต้องการวิธีการอัตโนมัติอย่างสมบูรณ์สำหรับคำอธิบายประกอบโครงสร้างของยีนที่แม่นยำ ด้วยเป้าหมายนี้ในใจเราได้พัฒนา Braker1 R1 R0 ซึ่งเป็นการรวมกันของ Genemark-ET R2 และ Augustus R3, R4 ที่ใช้ข้อมูลจีโนมและ RNA-seq เพื่อสร้างคำอธิบายประกอบโครงสร้างของยีนเต็มรูปแบบโดยอัตโนมัติในจีโนมนวนิยาย
อย่างไรก็ตามคุณภาพของข้อมูล RNA-seq ที่มีให้สำหรับการใส่คำอธิบายประกอบจีโนมใหม่เป็นตัวแปรและในบางกรณีข้อมูล RNA-seq ไม่สามารถใช้ได้เลย
Braker2 เป็นส่วนขยายของ Braker1 ซึ่งช่วยให้ การฝึกอบรมอัตโนมัติอย่างสมบูรณ์ ของเครื่องมือการทำนายยีน Genemark-ES/ET/EP/ETP R14, R15, R17, F1 และ Augustus จาก RNA-seq และ/หรือโปรตีน homology หลักฐานภายนอกจาก RNA-seq และข้อมูล homology โปรตีนใน การทำนาย
ตรงกันข้ามกับวิธีการอื่น ๆ ที่มีอยู่ซึ่งพึ่งพาข้อมูล homology โปรตีน Braker2 ถึงความแม่นยำในการทำนายของยีนสูงแม้ในกรณีที่ไม่มีคำอธิบายประกอบของสายพันธุ์ที่เกี่ยวข้องอย่างใกล้ชิดและในกรณีที่ไม่มีข้อมูล RNA-seq
Braker3 เป็นไปป์ไลน์ล่าสุดในชุด Braker มันช่วยให้การใช้ข้อมูล RNA-seq และ โปรตีนในท่ออัตโนมัติเต็มรูปแบบเพื่อฝึกอบรมและทำนายยีนที่เชื่อถือได้สูงด้วย Genemark-ETP และ Augustus ผลลัพธ์ของไปป์ไลน์คือชุดยีนรวมของเครื่องมือการทำนายยีนทั้งสองซึ่งมีเฉพาะยีนที่ได้รับการสนับสนุนสูงมากจากหลักฐานภายนอก
ในคู่มือผู้ใช้นี้เราจะอ้างถึง Braker1, Braker2 และ Braker3 เพียงแค่เป็น Braker เพราะพวกเขาถูกดำเนินการโดยสคริปต์เดียวกัน ( braker.pl )
ใช้ชุดจีโนมคุณภาพสูง หากคุณมีโครงนั่งร้านสั้น ๆ จำนวนมากในชุดจีโนมของคุณนั่งร้านสั้น ๆ เหล่านั้นจะเพิ่มรันไทม์อย่างมาก แต่จะไม่เพิ่มความแม่นยำในการทำนาย
ใช้ชื่อนั่งร้านอย่างง่ายในไฟล์จีโนม (เช่น >contig1 จะทำงานได้ดีกว่า >contig1my custom species namesome putative function /more/information/ and lots of special characters %&!*(){} ) ทำให้ชื่อนั่งร้านในไฟล์ Fasta ทั้งหมดของคุณง่ายขึ้นก่อนที่จะเรียกใช้โปรแกรมการจัดตำแหน่งใด ๆ
เพื่อที่จะทำนายยีนอย่างแม่นยำในจีโนมใหม่จีโนมควรถูกสวมหน้ากากเพื่อทำซ้ำ สิ่งนี้จะหลีกเลี่ยงการทำนายโครงสร้างของยีนบวกเท็จในภูมิภาคที่ซ้ำซากและมีความซับซ้อนต่ำ การปิดบังซ้ำก็เป็นสิ่งจำเป็นสำหรับการทำแผนที่ข้อมูล RNA-seq กับจีโนมด้วยเครื่องมือบางอย่าง (แม็ปเปอร์ RNA-seq อื่น ๆ เช่น HISAT2 ไม่สนใจข้อมูลการปิดบัง) ในกรณีของ Genemark-ES/ET/EP/ETP และ Augustus, Softmasking (เช่นการวางภูมิภาคซ้ำในตัวอักษรตัวพิมพ์เล็กและภูมิภาคอื่น ๆ ทั้งหมดลงในตัวอักษรตัวพิมพ์ใหญ่) นำไปสู่ผลลัพธ์ที่ดีกว่า hardmasking (เช่นการเปลี่ยนตัวอักษรในภูมิภาคซ้ำ ๆ N สำหรับนิวคลีโอไทด์ที่ไม่รู้จัก)
จีโนมจำนวนมากมีโครงสร้างของยีนที่จะทำนายอย่างถูกต้องด้วยพารามิเตอร์มาตรฐานของ Genemark-ES/ET/EP/ETP และ Augustus ภายใน Braker อย่างไรก็ตามจีโนมบางตัวมีคุณสมบัติเฉพาะ clade เช่นโมเดลจุดสาขาพิเศษในเชื้อราหรือรูปแบบไซต์ประกบที่ไม่ได้มาตรฐาน โปรดอ่านส่วนตัวเลือก [ตัวเลือก] เพื่อตรวจสอบว่าตัวเลือกใด ๆ ที่กำหนดเองอาจปรับปรุงความแม่นยำในการทำนายของยีนในจีโนมของสายพันธุ์เป้าหมายของคุณหรือไม่
ตรวจสอบผลการทำนายยีนเสมอก่อนการใช้งานเพิ่มเติม! คุณสามารถใช้เบราว์เซอร์จีโนมสำหรับการตรวจสอบด้วยภาพของแบบจำลองยีนในบริบทที่มีข้อมูลหลักฐานภายนอก Braker รองรับการสร้างฮับข้อมูลแทร็กสำหรับเบราว์เซอร์จีโนม UCSC ด้วย MakeHub เพื่อจุดประสงค์นี้
เบเกอร์ส่วนใหญ่มีข้อมูลกึ่งไม่ได้รับข้อมูลภายนอก (RNA-seq และ/หรือข้อมูลการจัดตำแหน่งโปรตีน spliced) สนับสนุนการฝึกอบรมของ Genemark-ES/ET/EP/ETP [F1] และการฝึกอบรมที่ตามมาของออกัสตัสกับการรวมหลักฐานภายนอก ขั้นตอนการทำนายยีน อย่างไรก็ตามขณะนี้มีท่อส่งเพิ่มเติมจำนวนหนึ่งที่รวมอยู่ใน Braker ในต่อไปนี้เราให้ภาพรวมของไฟล์อินพุตและท่อที่เป็นไปได้:
![braker2-main-a [รูปที่ 1]](https://images.downcodes.com/uploads/20250214/img_67aee79a0eaf534.png)
รูปที่ 2: Braker Pipeline A: การฝึกอบรม Genemark-ES บนข้อมูลจีโนมเท่านั้น; การทำนายยีน ab initio withaugustus
![braker2-main-b [รูปที่ 2]](https://images.downcodes.com/uploads/20250214/img_67aee79a0f13f35.png)
รูปที่ 3: Braker Pipeline B: การฝึกอบรม Genemark-ET ได้รับการสนับสนุนโดยข้อมูลการจัดตำแหน่ง RNA-seq spliced, การทำนายกับ Augustus ด้วยข้อมูลการจัดตำแหน่งแบบเดียวกัน
![braker2-main-c [รูปที่ 3]](https://images.downcodes.com/uploads/20250214/img_67aee79a0fa1036.png)
รูปที่ 4: Braker Pipeline C: การฝึกอบรม Genemark-EP+ ในการจัดตำแหน่งโปรตีน spliced, เริ่มต้นและหยุดข้อมูลการทำนายกับ Augustus ด้วยข้อมูลเดียวกันนั้นนอกจากนี้ยังมีคำแนะนำ CDSPART ที่ถูกล่ามโซ่ โปรตีนที่ใช้ในที่นี้อาจเป็นระยะทางวิวัฒนาการใด ๆ กับสิ่งมีชีวิตเป้าหมาย
![braker3-main-a [รูปที่ 4]](https://images.downcodes.com/uploads/20250214/img_67aee79a1010b37.png)
รูปที่ 5: Braker Pipeline D: หากจำเป็นให้ดาวน์โหลดและการจัดตำแหน่งของชุด RNA-seq สำหรับสายพันธุ์เป้าหมาย การฝึกอบรมของ Genemark-ETP ที่ได้รับการสนับสนุนโดยการจัดตำแหน่ง RNA-seq และฐานข้อมูลโปรตีนขนาดใหญ่ (โปรตีนสามารถอยู่ในระยะทางวิวัฒนาการใด ๆ ) ต่อจากนั้นการฝึกอบรมและการทำนายออกัสตัสโดยใช้ข้อมูลภายนอกเดียวกันพร้อมกับผลลัพธ์ของ Genemark-ETP การทำนายครั้งสุดท้ายคือการรวมกันของ TSEBRA ของผลลัพธ์ Augustus และ Genemark-ETP
เราตระหนักดีว่าการติดตั้ง "คู่มือ" ของ Braker3 และการพึ่งพาทั้งหมดนั้นน่าเบื่อและท้าทายจริงๆโดยไม่ได้รับอนุญาตจากราก ดังนั้นเราจึงจัดให้มีคอนเทนเนอร์ Docker ที่ได้รับการพัฒนาให้ทำงานด้วยความเป็นเอกเทศ ข้อมูลทั้งหมดเกี่ยวกับคอนเทนเนอร์นี้สามารถดูได้ที่ https://hub.docker.com/r/teambraker/braker3
ในระยะสั้นสร้างมันดังนี้:
singularity build braker3.sif docker://teambraker/braker3:latest
ดำเนินการด้วย:
singularity exec braker3.sif braker.pl
ทดสอบกับ:
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test1.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test2.sh .
singularity exec -B $PWD:$PWD braker3.sif cp /opt/BRAKER/example/singularity-tests/test3.sh .
export BRAKER_SIF=/your/path/to/braker3.sif # may need to modify
bash test1.sh
bash test2.sh
bash test3.sh
ผู้ใช้เพียงไม่กี่คนที่ต้องการเรียกใช้การวิเคราะห์ภายใน Docker (เนื่องจากต้องมีการอนุญาตรูท) อย่างไรก็ตามหากเป็นเป้าหมายของคุณคุณสามารถเรียกใช้และทดสอบคอนเทนเนอร์ดังนี้
sudo docker run --user 1000:100 --rm -it teambraker/braker3:latest bash
bash /opt/BRAKER/example/docker-tests/test1.sh # BRAKER1
bash /opt/BRAKER/example/docker-tests/test2.sh # BRAKER2
bash /opt/BRAKER/example/docker-tests/test3.sh # BRAKER3
ขอให้โชคดี ;-)
$PATH ของคุณอาจนำไปสู่การแทรกแซงที่ไม่คาดฝันทำให้โปรแกรมล้มเหลว โปรดย้ายรุ่น Genemark รุ่นเก่าทั้งหมดออกจาก $PATH ของคุณ (เช่น Genemark ใน ProtHint/dependencies )
ในช่วงเวลาของการเปิดตัวรุ่นเบเกอร์นี้ได้รับการทดสอบด้วย:
Augustus 3.5.0 F2
Genemark-ETP (แหล่งที่มาดู DockerFile)
Bamtools 2.5.1 R5
Samtools 1.7-4-G93586ED R6
Spaln 2.3.3d R8, R9, R10
NCBI Blast+ 2.2.31+ R12, R13
เพชร 0.9.24
Cdbfasta 0.99
Cdbyank 0.981
gushr 1.0.0
SRA Toolkit 3.00 R14
HISAT2 2.2.1 R15
Bedtools 2.30 R16
StringTie2 2.2.1 R17
GFFREAD 0.12.7 R18
รายงาน 0.2.5 R27
Braker ที่ใช้งานต้องใช้ระบบ Linux ด้วย bash และ Perl นอกจากนี้ Braker ยังต้องติดตั้งโมดูล CPAN-PERL ต่อไปนี้:
File::Spec::Functions
Hash::Merge
List::Util
MCE::Mutex
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
Math::Utils
File::HomeDir
สำหรับ Genemark-ETP ใช้เมื่อมีการจัดหาโปรตีนและ RNA-seq:
YAML::XSData::DumperThread::Queuethreads ตัวอย่างเช่น Ubuntu ติดตั้งโมดูลด้วย cpanminus F4 : sudo cpanm Module::Name เช่น sudo cpanm Hash::Merge
Braker ยังใช้โมดูล Perl helpMod_braker.pm ที่ไม่สามารถใช้ได้กับ CPAN โมดูลนี้เป็นส่วนหนึ่งของการเปิดตัวเบเกอร์และไม่จำเป็นต้องมีการติดตั้งแยกต่างหาก
หากคุณไม่มีสิทธิ์รูทบนเครื่อง Linux ลองตั้งค่า Anaconda (https://www.anaconda.com/distribution/) สภาพแวดล้อมดังนี้:
wget https://repo.anaconda.com/archive/Anaconda3-2018.12-Linux-x86_64.sh
bash bin/Anaconda3-2018.12-Linux-x86_64.sh # do not install VS (needs root privileges)
conda install -c anaconda perl
conda install -c anaconda biopython
conda install -c bioconda perl-app-cpanminus
conda install -c bioconda perl-file-spec
conda install -c bioconda perl-hash-merge
conda install -c bioconda perl-list-util
conda install -c bioconda perl-module-load-conditional
conda install -c bioconda perl-posix
conda install -c bioconda perl-file-homedir
conda install -c bioconda perl-parallel-forkmanager
conda install -c bioconda perl-scalar-util-numeric
conda install -c bioconda perl-yaml
conda install -c bioconda perl-class-data-inheritable
conda install -c bioconda perl-exception-class
conda install -c bioconda perl-test-pod
conda install -c bioconda perl-file-which # skip if you are not comparing to reference annotation
conda install -c bioconda perl-mce
conda install -c bioconda perl-threaded
conda install -c bioconda perl-list-util
conda install -c bioconda perl-math-utils
conda install -c bioconda cdbtools
conda install -c eumetsat perl-yaml-xs
conda install -c bioconda perl-data-dumper
ต่อมาติดตั้งเบเกอร์และซอฟต์แวร์อื่น ๆ "ตามปกติ" ในขณะที่อยู่ในสภาพแวดล้อม conda ของคุณ หมายเหตุ: มีแพ็คเกจ Bioconda Braker และแพ็คเกจ Bioconda Augustus พวกเขาทำงาน แต่พวกเขามักจะล้าหลังรหัสการพัฒนาของเครื่องมือทั้งสองบน GitHub ดังนั้นเราจึงแนะนำการติดตั้งด้วยตนเองและการใช้แหล่งข้อมูลล่าสุด
Braker เป็นคอลเลกชันของสคริปต์ Perl และ Python และโมดูล Perl สคริปต์หลักที่จะถูกเรียกเพื่อเรียกใช้เบเกอร์คือ braker.pl ส่วนประกอบ Perl และ Python เพิ่มเติมคือ:
align2hints.pl
filterGenemark.pl
filterIntronsFindStrand.pl
startAlign.pl
helpMod_braker.pm
findGenesInIntrons.pl
downsample_traingenes.pl
ensure_n_training_genes.py
get_gc_content.py
get_etp_hints.py
สคริปต์ทั้งหมด (ไฟล์ที่ลงท้ายด้วย *.pl และ *.py ) ที่เป็นส่วนหนึ่งของเบเกอร์จะต้องดำเนินการเพื่อเรียกใช้เบเกอร์ นี่ควรเป็นกรณีนี้หากคุณดาวน์โหลด Braker จาก GitHub ความสามารถในการปฏิบัติงานอาจถูกเขียนทับหากคุณถ่ายโอนเบเกอร์บน USB-stick ไปยังคอมพิวเตอร์เครื่องอื่น เพื่อตรวจสอบว่าไฟล์ที่จำเป็นสามารถเรียกใช้งานได้ให้เรียกใช้คำสั่งต่อไปนี้ในไดเรกทอรีที่มีสคริปต์ Braker Perl:
ls -l *.pl *.py
เอาต์พุตควรคล้ายกับนี้:
-rwxr-xr-x 1 katharina katharina 18191 Mai 7 10:25 align2hints.pl
-rwxr-xr-x 1 katharina katharina 6090 Feb 19 09:35 braker_cleanup.pl
-rwxr-xr-x 1 katharina katharina 408782 Aug 17 18:24 braker.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:25 downsample_traingenes.pl
-rwxr-xr-x 1 katharina katharina 5024 Mai 7 10:23 ensure_n_training_genes.py
-rwxr-xr-x 1 katharina katharina 4542 Apr 3 2019 filter_augustus_gff.pl
-rwxr-xr-x 1 katharina katharina 30453 Mai 7 10:25 filterGenemark.pl
-rwxr-xr-x 1 katharina katharina 5754 Mai 7 10:25 filterIntronsFindStrand.pl
-rwxr-xr-x 1 katharina katharina 7765 Mai 7 10:25 findGenesInIntrons.pl
-rwxr-xr-x 1 katharina katharina 1664 Feb 12 2019 gatech_pmp2hints.pl
-rwxr-xr-x 1 katharina katharina 2250 Jan 9 13:55 log_reg_prothints.pl
-rwxr-xr-x 1 katharina katharina 4679 Jan 9 13:55 merge_transcript_sets.pl
-rwxr-xr-x 1 katharina katharina 41674 Mai 7 10:25 startAlign.pl
เป็นสิ่งสำคัญที่ x ใน -rwxr-xr-x จะปรากฏขึ้นสำหรับแต่ละสคริปต์ หากไม่เป็นเช่นนั้นให้เรียกใช้
`chmod a+x *.pl *.py`
เพื่อเปลี่ยนแอตทริบิวต์ไฟล์
คุณอาจพบว่ามีประโยชน์ในการเพิ่มไดเรกทอรีที่สคริปต์ Perl Perl อยู่ในตัวแปรสภาพแวดล้อม $PATH ของคุณ สำหรับเซสชันทุบตีครั้งเดียวป้อน:
PATH=/your_path_to_braker/:$PATH
export PATH
เพื่อให้การปรับเปลี่ยน $PATH นี้พร้อมใช้งานสำหรับเซสชัน Bash ทั้งหมดให้เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc )
Braker เรียกใช้เครื่องมือซอฟต์แวร์ชีวสารสนเทศต่างๆที่ไม่ได้เป็นส่วนหนึ่งของ Braker เครื่องมือบางอย่างมีข้อผูกมัดเช่น Braker จะไม่ทำงานเลยหากเครื่องมือเหล่านี้ไม่ได้อยู่ในระบบของคุณ เครื่องมืออื่น ๆ เป็นทางเลือก โปรดติดตั้งเครื่องมือทั้งหมดที่จำเป็นสำหรับการใช้งาน Braker ในโหมดที่คุณเลือก
ดาวน์โหลด genemark-etp f1 จาก http://github.com/gatech-genemark/genemark-etp หรือ https://topaz.gatech.edu/genemark/etp.for_braker.tar.gz คลายและติดตั้ง Genemark-ETP ตามที่อธิบายไว้ในไฟล์ README ของ Genemark-ETP
หากมีอยู่ในตัวแปร $PATH ของคุณแล้ว Braker จะเดาตำแหน่งของ gmes_petap.pl หรือ gmetp.pl โดยอัตโนมัติ มิฉะนั้น Braker สามารถค้นหา Genemark-ES/ET/EP/ETP Executables ได้โดยการค้นหาในตัวแปรสภาพแวดล้อม GENEMARK_PATH หรือโดยใช้อาร์กิวเมนต์บรรทัดคำสั่ง ( --GENEMARK_PATH=/your_path_to_GeneMark_executables/ )
ในการตั้งค่าตัวแปรสภาพแวดล้อมสำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
export GENEMARK_PATH=/your_path_to_GeneMark_executables/
เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อให้สามารถใช้งานได้สำหรับเซสชันทุบตีทั้งหมด
สคริปต์ Perl ภายใน Genemark-ES/ET/EP/ETP ได้รับการกำหนดค่าด้วยตำแหน่ง Perl เริ่มต้น AT /usr/bin/perl
หากคุณใช้งาน GenEmark-ES/ET/EP/ETP ในสภาพแวดล้อม Anaconda (หรือต้องการใช้ Perl จากตัวแปร $PATH ด้วยเหตุผลอื่นใด) ให้ปรับเปลี่ยน Shebang ของสคริปต์ Genemark-ES/ET/EP/ETP ทั้งหมดด้วย คำสั่งต่อไปนี้อยู่ในโฟลเดอร์ Genemark-ES/ET/EP/ETP:
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
คุณสามารถตรวจสอบว่า Genemark-ES/ET/EP ได้รับการติดตั้งอย่างถูกต้องหรือไม่โดยเรียกใช้ check_install.bash และ/หรือการดำเนินการตัวอย่างในไดเรกทอรี GeneMark-E-tests
Genemark-ETP นั้นเข้ากันได้กับลงมาเช่นมันครอบคลุมการทำงานของ Genemark-EP และ Genemark-ET ใน Braker เช่นกัน
ดาวน์โหลดออกัสตัสจากสาขาหลักที่ https://github.com/gaius-augustus/augustus แกะออกัสตัสและติดตั้งออกัสตัสตาม Augustus README.TXT อย่าใช้เวอร์ชันออกัสตัสที่ล้าสมัยจากแหล่งอื่น ๆ เช่นแพ็คเกจ Debian หรือแพ็คเกจ Bioconda! เบเกอร์ขึ้นอยู่กับโดยเฉพาะอย่างยิ่งในไดเรกทอรีออกัสตัส/สคริปต์ที่ทันสมัยและแหล่งข้อมูลอื่น ๆ มักจะล้าหลัง
คุณควรรวบรวมออกัสตัสในระบบของคุณเองเพื่อหลีกเลี่ยงปัญหาเกี่ยวกับห้องสมุดรุ่นที่ออกัสตัสใช้ คำแนะนำในการรวบรวมมีให้ในไฟล์ Augustus README.TXT ( Augustus/README.txt )
ออกัสตัสประกอบด้วย augustus เครื่องมือทำนายยีนเครื่องมือ C ++ เพิ่มเติมที่อยู่ใน Augustus/auxprogs และสคริปต์ Perl ที่ตั้งอยู่ใน Augustus/scripts สคริปต์ Perl จะต้องสามารถเรียกใช้งานได้ (ดูคำแนะนำในส่วนของส่วนประกอบเบเกอร์
เครื่องมือ C ++ bam2hints เป็นองค์ประกอบสำคัญของเบเกอร์เมื่อทำงานด้วย RNA-seq แหล่งที่มาตั้งอยู่ใน Augustus/auxprogs/bam2hints ตรวจสอบให้แน่ใจว่าคุณรวบรวม bam2hints ในระบบของคุณ (ควรรวบรวมโดยอัตโนมัติเมื่อ Augustus ถูกรวบรวม แต่ในกรณีที่มีปัญหากับ bam2hints โปรดอ่านคำแนะนำในการแก้ไขปัญหาใน Augustus/auxprogs/bam2hints/README )
เนื่องจาก Braker เป็นไปป์ไลน์ที่ฝึกอบรมออกัสตัสเช่นเขียนไฟล์พารามิเตอร์เฉพาะสปีชีส์เบเกอร์จึงต้องเขียนการเข้าถึงไดเรกทอรีการกำหนดค่าของออกัสตัสที่มีไฟล์ดังกล่าว ( Augustus/config/ ) หากคุณติดตั้ง Augustus ทั่วโลกในระบบของคุณโดยทั่วไปโฟลเดอร์ config จะไม่สามารถเขียนได้โดยผู้ใช้ทุกคน ไม่ว่าจะเป็นไดเรกทอรีที่การกำหนด config อยู่ที่ผู้ใช้ของออกัสตัสหรือคัดลอก config/ โฟลเดอร์ (ซ้ำ) ไปยังสถานที่ที่ผู้ใช้ได้รับอนุญาตเขียน
ออกัสตัสจะค้นหาโฟลเดอร์ config โดยมองหาตัวแปรสภาพแวดล้อม $AUGUSTUS_CONFIG_PATH หากไม่ได้ตั้งค่าตัวแปรสภาพแวดล้อม $AUGUSTUS_CONFIG_PATH แล้วเบเกอร์จะมองในเส้นทาง ../config เทียบกับไดเรกทอรีที่พบว่ามีการดำเนินการของออกัสตัส หรือคุณสามารถจัดหาตัวแปรเป็นอาร์กิวเมนต์บรรทัดคำสั่งให้กับเบเกอร์ ( --AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/ ) เราขอแนะนำให้คุณส่งออกตัวแปรเช่นสำหรับเซสชันการทุบตีปัจจุบันของคุณ:
export AUGUSTUS_CONFIG_PATH=/your_path_to_AUGUSTUS/Augustus/config/
เพื่อให้ตัวแปรพร้อมใช้งานสำหรับเซสชันทุบตีทั้งหมดให้เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้นเช่น ~/.bashrc
โปรดดูที่ DockerFile ในกรณีที่คุณต้องการติดตั้ง Augustus เป็นแพ็คเกจ Debian จำนวนสคริปต์ต้องได้รับการแก้ไขแล้ว
Braker คาดว่าไดเรกทอรี config ทั้งหมดของออกัสตัสที่ $AUGUSTUS_CONFIG_PATH เช่น species ปีชีส์ย่อยโฟลเดอร์ที่มีเนื้อหา (อย่างน้อย generic ) และ extrinsic ! การจัดทำโฟลเดอร์ที่เขียนได้ แต่ว่างเปล่าที่ $AUGUSTUS_CONFIG_PATH จะไม่ทำงานให้กับ Braker หากคุณต้องการแยก Augustus Binary และ $AUGUSTUS_CONFIG_PATH เราขอแนะนำให้คุณคัดลอกเนื้อหาการกำหนดค่าที่ไม่สามารถเขียนได้ซ้ำไปยังตำแหน่งที่เขียนได้
หากคุณมีการติดตั้ง Augustus AT /usr/bin/augustus ทั่วทั้งระบบทั่วทั้งระบบจะมีสำเนา config ที่ไม่สามารถแก้ไขได้อยู่ที่ /usr/bin/augustus_config/ โฟลเดอร์ /home/yours/ เขียนได้สำหรับคุณ คัดลอกด้วยคำสั่งต่อไปนี้ (และตั้งค่าตัวแปรที่ต้องการเพิ่มเติม):
cp -r /usr/bin/Augustus/config/ /home/yours/
export AUGUSTUS_CONFIG_PATH=/home/yours/augustus_config
export AUGUSTUS_BIN_PATH=/usr/bin
export AUGUSTUS_SCRIPTS_PATH=/usr/bin/augustus_scripts
การเพิ่มไดเรกทอรีของไบนารีออกัสตัสและสคริปต์ลงในตัวแปร $PATH ของคุณช่วยให้ระบบของคุณสามารถค้นหาเครื่องมือเหล่านี้โดยอัตโนมัติ มันไม่ได้เป็นข้อกำหนดสำหรับการใช้งาน Braker ในการทำ $AUGUSTUS_CONFIG_PATH นี้เพราะ braker.pl เพิ่มลงในตัวแปร $PATH ของคุณ สำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
PATH=:/your_path_to_augustus/bin/:/your_path_to_augustus/scripts/:$PATH
export PATH
สำหรับเซสชันทุบตีทั้งหมดของคุณเพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc )
ใน Ubuntu Python3 มักจะติดตั้งโดยค่าเริ่มต้น python3 จะอยู่ในตัวแปร $PATH ของคุณโดยค่าเริ่มต้นและ Braker จะค้นหาโดยอัตโนมัติ อย่างไรก็ตามคุณมีตัวเลือกในการระบุตำแหน่งไบนารี python3 ในอีกสองวิธี:
ส่งออกตัวแปรสภาพแวดล้อม $PYTHON3_PATH เช่นในไฟล์ ~/.bashrc ของคุณ:
export PYTHON3_PATH=/path/to/python3/
ระบุตัวเลือกบรรทัดคำสั่ง --PYTHON3_PATH=/path/to/python3/ to braker.pl
ดาวน์โหลด bamtools (เช่น git clone https://github.com/pezmaster31/bamtools.git ) ติดตั้ง BAMTools โดยพิมพ์ต่อไปนี้ในเปลือกของคุณ:
cd your-bamtools-directory mkdir build cd build cmake .. make
หากอยู่ในตัวแปร $PATH ของคุณแล้วเบเกอร์จะพบ Bamtools โดยอัตโนมัติ มิฉะนั้น Braker สามารถค้นหา Bamtools Binary ได้โดยใช้ตัวแปรสภาพแวดล้อม $BAMTOOLS_PATH หรือโดยใช้อาร์กิวเมนต์บรรทัดคำสั่ง ( --BAMTOOLS_PATH=/your_path_to_bamtools/bin/ f6 ) ในการตั้งค่าตัวแปรสภาพแวดล้อมเช่นสำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
export BAMTOOLS_PATH=/your_path_to_bamtools/bin/
เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อตั้งค่าตัวแปรสภาพแวดล้อมสำหรับเซสชันทุบตีทั้งหมด
คุณสามารถใช้ NCBI BLAST+ หรือ Diamond เพื่อกำจัดยีนฝึกอบรมซ้ำซ้อน คุณไม่ต้องการเครื่องมือทั้งสอง หากเพชรอยู่จะเป็นที่ต้องการเพราะมันเร็วกว่ามาก
ได้รับและแกะเพชรออกดังนี้:
wget http://github.com/bbuchfink/diamond/releases/download/v0.9.24/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
หากอยู่ในตัวแปร $PATH ของคุณแล้ว Braker จะพบ Diamond โดยอัตโนมัติ มิฉะนั้นเบเกอร์สามารถค้นหาเพชรไบนารีได้โดยใช้ตัวแปรสภาพแวดล้อม $DIAMOND_PATH หรือโดยการโต้แย้งบรรทัดคำสั่ง ( --DIAMOND_PATH=/your_path_to_diamond ) ในการตั้งค่าตัวแปรสภาพแวดล้อมเช่นสำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
export DIAMOND_PATH=/your_path_to_diamond/
เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อตั้งค่าตัวแปรสภาพแวดล้อมสำหรับเซสชันทุบตีทั้งหมด
หากคุณตัดสินใจใช้ BLAST+ ให้ติดตั้ง NCBI Blast+ ด้วย sudo apt-get install ncbi-blast+
หากอยู่ในตัวแปร $PATH ของคุณแล้ว Braker จะพบ Blastp โดยอัตโนมัติ มิฉะนั้น Braker สามารถค้นหา BLASTP Binary ได้โดยใช้ตัวแปรสภาพแวดล้อม $BLAST_PATH หรือโดยใช้อาร์กิวเมนต์บรรทัดคำสั่ง ( --BLAST_PATH=/your_path_to_blast/ ) ในการตั้งค่าตัวแปรสภาพแวดล้อมเช่นสำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
export BLAST_PATH=/your_path_to_blast/
เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อตั้งค่าตัวแปรสภาพแวดล้อมสำหรับเซสชันทุบตีทั้งหมด
เครื่องมือต่อไปนี้เป็นสิ่งจำเป็นโดย GenEmark-ETP และจะพยายามค้นหาในตัวแปร $PATH ของคุณ ดังนั้นให้แน่ใจว่าได้เพิ่มตำแหน่งของพวกเขาใน $PATH ของคุณเช่น:
export PATH=$PATH:/your/path/to/Tool
สำหรับเครื่องมือทั้งหมดด้านล่างให้เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อขยายตัวแปร $PATH ของคุณสำหรับเซสชัน Bash ทั้งหมด
เครื่องมือซอฟต์แวร์เหล่านี้เป็นสิ่งจำเป็นเฉพาะในกรณีที่คุณเรียกใช้ Braker ด้วยข้อมูล RNA-seq และ โปรตีน!
StringTie2 ถูกใช้โดย Genemark-ETP เพื่อประกอบการจัดตำแหน่ง RNA-seq ที่จัดตำแหน่ง สามารถดาวน์โหลด StringTie2 เวอร์ชัน preompiled ได้จาก https://ccb.jhu.edu/software/stringtie/#install
แพคเกจซอฟต์แวร์ Bedtools เป็นสิ่งจำเป็นโดย Genemark-ETP หากคุณต้องการเรียกใช้ Braker ด้วยข้อมูล RNA-seq และโปรตีน คุณสามารถดาวน์โหลด bedtools ได้จาก https://github.com/arq5x/bedtools2/releases ที่นี่คุณสามารถดาวน์โหลด bedtools.static.binary , eg เวอร์ชัน precompiled
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x
หรือคุณสามารถดาวน์โหลด bedtools-2.30.0.tar.gz และรวบรวมจากแหล่งที่มาโดยใช้ make เช่น
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools-2.30.0.tar.gz
tar -zxvf bedtools-2.30.0.tar.gz
cd bedtools2
make
ดู https://bedtools.readthedocs.io/en/latest/content/installation.html สำหรับข้อมูลเพิ่มเติม
GFFREAD เป็นซอฟต์แวร์ยูทิลิตี้ที่ต้องการโดย GenEmark-ETP สามารถดาวน์โหลดได้จาก make
wget https://github.com/gpertea/gffread/releases/download/v0.12.7/gffread-0.12.7.Linux_x86_64.tar.gz
tar xzf gffread-0.12.7.Linux_x86_64.tar.gz
cd gffread-0.12.7.Linux_x86_64
make
Samtools ไม่จำเป็นสำหรับการเรียกใช้ Braker โดยไม่ต้องใช้ Genemark-ETP หากไฟล์ทั้งหมดของคุณถูกจัดรูปแบบอย่างถูกต้อง (เช่นลำดับทั้งหมดควรมีชื่อ Fasta สั้นและไม่ซ้ำกัน) หากคุณไม่แน่ใจว่าไฟล์ทั้งหมดของคุณถูก fomatted อย่างถูกต้องอาจเป็นประโยชน์ในการติดตั้ง samtools เนื่องจากเบเกอร์สามารถแก้ไขปัญหารูปแบบได้โดยอัตโนมัติโดยใช้ samtools
เป็นข้อกำหนดเบื้องต้นสำหรับ samtools ดาวน์โหลดและติดตั้ง htslib (เช่น git clone https://github.com/samtools/htslib.git ทำตามเอกสาร htslib สำหรับการติดตั้ง)
ดาวน์โหลดและติดตั้ง samtools (เช่น git clone git://github.com/samtools/samtools.git ) ต่อมาตามเอกสาร Samtools สำหรับการติดตั้ง)
หากอยู่ในตัวแปร $PATH ของคุณแล้วเบเกอร์จะพบ samtools โดยอัตโนมัติ มิฉะนั้น Braker สามารถค้นหา samtools ได้โดยใช้อาร์กิวเมนต์บรรทัดคำสั่ง ( --SAMTOOLS_PATH=/your_path_to_samtools/ ) หรือโดยใช้ตัวแปรสภาพแวดล้อม $SAMTOOLS_PATH สำหรับการส่งออกตัวแปรเช่นสำหรับเซสชันทุบตีปัจจุบันของคุณพิมพ์:
export SAMTOOLS_PATH=/your_path_to_samtools/
เพิ่มบรรทัดด้านบนลงในสคริปต์เริ่มต้น (เช่น ~/.bashrc ) เพื่อตั้งค่าตัวแปรสภาพแวดล้อมสำหรับเซสชันทุบตีทั้งหมด
หากมีการติดตั้ง biopython เบเกอร์สามารถสร้างไฟล์ fasta ด้วยลำดับการเข้ารหัสและลำดับโปรตีนที่ทำนายโดยออกัสตัสและสร้างฮับข้อมูลแทร็กสำหรับการสร้างภาพของเบเกอร์ที่ทำงานด้วย makehub R16 นี่เป็นขั้นตอนเสริม ตัวแรกสามารถปิดใช้งานได้ด้วยธงบรรทัดคำสั่ง --skipGetAnnoFromFasta , ที่สองสามารถเปิดใช้งานได้โดยใช้ตัวเลือกบรรทัดคำสั่ง --makehub [email protected] ไม่จำเป็นต้องใช้ biopython จะต้องดำเนินการ
บน Ubuntu ติดตั้ง Python3 Package Manager ด้วย:
`sudo apt-get install python3-pip`
จากนั้นติดตั้ง biopython ด้วย:
`sudo pip3 install biopython`
CDBFASTA และ CDBYANK เป็นสิ่งจำเป็นโดย Braker สำหรับการแก้ไขยีนออกัสตัสด้วย in frame stop codons (spliced stop codons) โดยใช้สคริปต์ Augustus FIX_IN_FRAME_STOP_CODON_GENES.PY สิ่งนี้สามารถข้ามไปด้วย --skip_fixing_broken_genes
บน Ubuntu ติดตั้ง cdbfasta ด้วย:
sudo apt-get install cdbfasta
สำหรับระบบอื่น ๆ คุณสามารถรับ cdbfasta จาก https://github.com/gpertea/cdbfasta, เช่น:
git clone https://github.com/gpertea/cdbfasta.git
cd cdbfasta
make all
ใน Ubuntu, CDBFasta และ Cdbyank จะอยู่ในตัวแปร $PATH ของคุณหลังจากการติดตั้งและ Braker จะค้นหาโดยอัตโนมัติ อย่างไรก็ตามคุณมีตัวเลือกในการระบุที่ตั้ง cdbfasta และ cdbyank ไบนารีในอีกสองวิธี:
$CDBTOOLS_PATH เช่นในไฟล์ ~/.bashrc ของคุณ: export CDBTOOLS_PATH=/path/to/cdbtools/
--CDBTOOLS_PATH=/path/to/cdbtools/ to braker.pl หมายเหตุ: การสนับสนุนของสแตนด์อโลน spaln (ouside ของ prothint) ภายในเบเกอร์ถูกเลิกใช้
เครื่องมือนี้เป็นสิ่งจำเป็นหากคุณเรียกใช้ prothint หรือถ้าคุณต้องการเรียกใช้โปรตีนไปยังการจัดตำแหน่งจีโนมกับ Braker โดยใช้ spaln นอก Prothint การใช้ spaln นอก Prothint เป็นวิธีที่เหมาะสมเฉพาะในกรณีที่มีการเพิ่มความสามารถในการวิวัฒนาการระยะทางสั้น ๆ ของจีโนมเป้าหมายของคุณ เราขอแนะนำให้ใช้ Spaln ผ่าน Prothint สำหรับ Braker Prothint นำไบนารี่ spaln หากไม่ได้ผลในระบบของคุณให้ดาวน์โหลด Spaln จาก https://github.com/ogotoh/spaln แกะและติดตั้งตาม spaln/doc/SpalnReadMe22.pdf
Braker จะพยายามค้นหาการดำเนินการ spaln โดยใช้ตัวแปรสภาพแวดล้อม $ALIGNMENT_TOOL_PATH อีกทางเลือกหนึ่งสามารถจัดหาเป็นอาร์กิวเมนต์บรรทัดคำสั่ง ( --ALIGNMENT_TOOL_PATH=/your/path/to/spaln )
เครื่องมือนี้จำเป็นก็ต่อเมื่อคุณต้องการเพิ่ม UTR (จากข้อมูล RNA-seq) ไปยังยีนที่คาดการณ์หรือหากคุณต้องการฝึกอบรมพารามิเตอร์ UTR สำหรับออกัสตัสและทำนายยีนด้วย UTRS ไม่ว่าในกรณีใด Gushr ต้องการอินพุตของข้อมูล RNA-seq
Gushr สามารถดาวน์โหลดได้ที่ https://github.com/gaius-augustus/gushr รับมันโดยการพิมพ์:
git clone https://github.com/Gaius-Augustus/GUSHR.git
Gushr ดำเนินการไฟล์ Jar Gemoma R19, R20, R21 และไฟล์ JAR นี้ต้องใช้ Java 1.8 บน Ubuntu คุณสามารถติดตั้ง Java 1.8 ด้วยคำสั่งต่อไปนี้:
sudo apt-get install openjdk-8-jdk
หากคุณติดตั้ง Java หลายรุ่นในระบบของคุณตรวจสอบให้แน่ใจว่าคุณเปิดใช้งาน 1.8 Braker ก่อนการใช้งานด้วย Java โดยใช้งาน
sudo update-alternatives --config java
และเลือกเวอร์ชันที่ถูกต้อง
หากคุณสลับ --UTR=on , bamtowig.py จะต้องใช้เครื่องมือต่อไปนี้ที่สามารถดาวน์โหลดได้จาก http://hgdownload.soe.ucsc.edu/admin/exe:
twobitinfo
fatotwobit
เป็นทางเลือกในการติดตั้งเครื่องมือเหล่านี้ลงในเส้นทาง $ ของคุณ หากคุณไม่ทำและคุณสลับ --UTR=on , bamtowig.py จะดาวน์โหลดโดยอัตโนมัติลงในไดเรกทอรีการทำงาน
หากคุณต้องการสร้างฮับข้อมูลแทร็กของ Braker โดยอัตโนมัติซอฟต์แวร์ MakeHub ที่มีอยู่ที่ https://github.com/gaius-augustus/makehub เป็นสิ่งจำเป็น ดาวน์โหลดซอฟต์แวร์ (ไม่ว่าจะโดยใช้ git clone https://github.com/Gaius-Augustus/MakeHub.git หรือโดยเลือกการเปิดตัวจาก https://github.com/gaius-augustus/makehub/releases แพ็คเกจหากคุณดาวน์โหลดรีลีส (เช่น unzip MakeHub.zip หรือ tar -zxvf MakeHub.tar.gz
เบเกอร์จะพยายามค้นหาสคริปต์ make_hub.py โดยใช้ตัวแปรสภาพแวดล้อม $MAKEHUB_PATH อีกทางเลือกหนึ่งนี้สามารถจัดหาเป็นอาร์กิวเมนต์บรรทัดคำสั่ง ( --MAKEHUB_PATH=/your/path/to/MakeHub/ ) เบเกอร์ยังสามารถลองเดาตำแหน่งของ MakeHub ในระบบของคุณ
หากคุณต้องการให้ Braker ดาวน์โหลดไลบรารี RNA-seq จาก SRA ของ NCBI จำเป็นต้องใช้ชุดเครื่องมือ SRA คุณสามารถรับชุดเครื่องมือ SRA รุ่น precompiled ได้จาก http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221
Braker จะพยายามค้นหาไบนารีที่ดำเนินการได้จากชุดเครื่องมือ SRA (FastQ-Dump, Prefetch) โดยใช้ตัวแปรสภาพแวดล้อม $SRATOOLS_PATH อีกทางเลือกหนึ่งสามารถจัดหาเป็นอาร์กิวเมนต์บรรทัดคำสั่ง ( --SRATOOLS_PATH=/your/path/to/SRAToolkit/ ) เบเกอร์ยังสามารถลองเดาตำแหน่งของชุดเครื่องมือ SRA บนระบบของคุณได้หากผู้ดำเนินการอยู่ในตัวแปร $PATH ของคุณ
หากคุณต้องการใช้การอ่าน RNA-seq ที่ไม่ได้จัดตำแหน่งซอฟต์แวร์ HISAT2 จะต้องทำแผนที่กับจีโนม สามารถดาวน์โหลด HISAT2 เวอร์ชัน preompiled ได้จาก http://daehwankimlab.github.io/hisat2/download/#version-hisat2-221
Braker จะพยายามค้นหา HISAT2 Binaries (HISAT2, HISAT2-BUILD) โดยใช้ตัวแปรสภาพแวดล้อม $HISAT2_PATH อีกทางเลือกหนึ่งสามารถจัดหาเป็นอาร์กิวเมนต์บรรทัดคำสั่ง ( --HISAT2_PATH=/your/path/to/HISAT2/ ) Braker ยังสามารถลองเดาตำแหน่งของ HISAT2 ในระบบของคุณได้หาก Executables อยู่ในตัวแปร $PATH ของคุณ
หากคุณต้องการเรียกใช้ TSEBRA ภายใน Braker ในโหมดการเพิ่มความสมบูรณ์ของ BUSCO คุณต้องติดตั้งการตอบสนอง
wget https://github.com/huangnengCSU/compleasm/releases/download/v0.2.4/compleasm-0.2.4_x64-linux.tar.bz2
tar -xvjf compleasm-0.2.4_x64-linux.tar.bz2 &&
เพิ่มผลลัพธ์ของโฟลเดอร์ที่ได้รับในตัวแปร $PATH ของคุณเช่น:
export PATH=$PATH:/your/path/to/compleasm_kit
การตอบสนองต้องใช้แพนด้าซึ่งสามารถติดตั้งได้ด้วย:
pip install pandas
Braker (Braker.PL) ใช้ GetConf เพื่อดูว่ามีกี่เธรดที่สามารถทำงานบนระบบของคุณได้ บน Ubuntu คุณสามารถติดตั้งด้วย:
sudo apt-get install libc-bin
ในต่อไปนี้เราจะอธิบายถึงเบเกอร์“ ทั่วไป” เรียกร้องให้มีประเภทข้อมูลอินพุตที่แตกต่างกัน โดยทั่วไปเราขอแนะนำให้คุณเรียกใช้ Braker ในลำดับจีโนมที่ได้รับการทำซ้ำเพื่อทำซ้ำ เบเกอร์ควรนำไปใช้กับจีโนมที่ได้รับการทำซ้ำเพื่อทำซ้ำ!
This approach is suitable for genomes of species for which RNA-Seq libraries with good transcriptome coverage are available and for which protein data is not at hand. The pipeline is illustrated in Figure 2.
BRAKER has several ways to receive RNA-Seq data as input:
You can provide ID(s) of RNA-Seq libraries from SRA (in case of multiple IDs, separate them by comma) as argument to --rnaseq_sets_ids . The libraries belonging to the IDs are then downloaded automatically by BRAKER, eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
You can use local FASTQ file(s) of unaligned reads as input. In this case, you have to provide BRAKER with the ID(s) of the RNA-Seq set(s) as argument to --rnaseq_sets_ids and the path(s) to the directories, where the FASTQ files are located as argument to --rnaseq_sets_dirs . For each ID ID , BRAKER will search in these directories for one FASTQ file named ID.fastq if the reads are unpaired, or for two FASTQ files named ID_1.fastq and ID_2.fastq if they are paired.
For example, if you have a paired library called 'SRA_ID1' and an unpaired library named 'SRA_ID2', you have to have a directory /path/to/local/fastq/files/ , where the files SRA_ID1_1.fastq , SRA_ID1_2.fastq , and SRA_ID2.fastq reside. Then, you could run BRAKER with following command:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/fastq/files/
There are two ways of supplying BRAKER with RNA-Seq data as bam file(s). First, you can do it in the same way as you would supply FASTQ file(s): Provide the ID(s)/name(s) of your bam file(s) as argument to --rnaseq_sets_ids and specify directories where the bam files reside with --rnaseq_sets_dirs . BRAKER will automatically detect that these ID(s) are bam and not FASTQ file(s), eg:
braker.pl --species=yourSpecies --genome=genome.fasta
--rnaseq_sets_ids=BAM_ID1,BAM_ID2
--rnaseq_sets_dirs=/path/to/local/bam/files/
Second, you can specify the paths to your bam file(s) directly, eg can either extract RNA-Seq spliced alignment information from bam files, or it can use such extracted information, directly.
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file1.bam,file2.bam
Please note that we generally assume that bam files were generated with HiSat2 because that is the aligner that would also be executed by BRAKER3 with fastq input. If you want for some reason to generate the bam files with STAR, use the option --outSAMstrandField intronMotif of STAR to produce files that are compatible wiht StringTie in BRAKER3.
In order to run BRAKER with RNA-Seq spliced alignment information that has already been extracted, run:
braker.pl --species=yourSpecies --genome=genome.fasta
--hints=hints1.gff,hints2.gff
The format of such a hints file must be as follows (tabulator separated file):
chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=E
...
The source b2h in the second column and the source tag src=E in the last column are essential for BRAKER to determine whether a hint has been generated from RNA-Seq data.
It is also possible to provide RNA-Seq sets in different ways for the same BRAKER run, any combination of above options is possible. It is not recommended to provide RNA-Seq data with --hints if you run BRAKER in ETPmode (RNA-Seq and protein data), because GeneMark-ETP won't use these hints!
This approach is suitable for genomes of species for which no RNA-Seq libraries are available. A large database of proteins (with possibly longer evolutionary distance to the target species) should be used in this case. This mode is illustrated in figure 9.
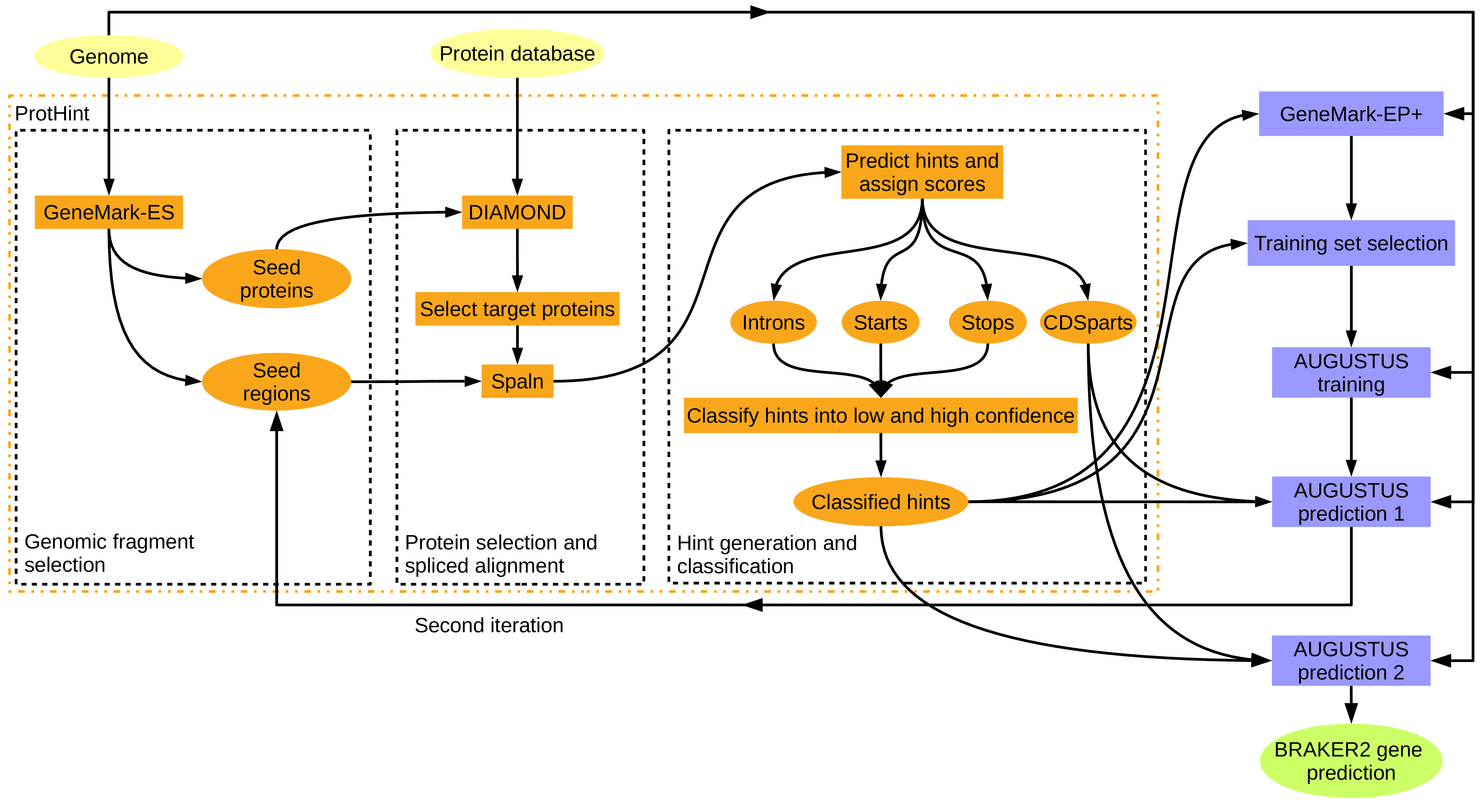
Figure 9: BRAKER with proteins of any evolutionary distance. ProtHint protein mapping pipelines is used to generate protein hints. ProtHint automatically determines which alignments are from close relatives, and which are from rather distant relatives.
For running BRAKER in this mode, type:
braker.pl --genome=genome.fa --prot_seq=proteins.fa
We recommend using OrthoDB as basis for proteins.fa . The instructions on how to prepare the input OrthoDB proteins are documented here: https://github.com/gatech-genemark/ProtHint#protein-database-preparation.
You can of course add additional protein sequences to that file, or try with a completely different database. Any database will need several representatives for each protein, though.
Instead of having BRAKER run ProtHint, you can also start BRAKER with hints already produced by ProtHint, by providing ProtHint's prothint_augustus.gff output:
braker.pl --genome=genome.fa --hints=prothint_augustus.gff
The format of prothint_augustus.gff in this mode looks like this:
2R ProtHint intron 11506230 11506648 4 + . src=M;mult=4;pri=4
2R ProtHint intron 9563406 9563473 1 + . grp=69004_0:001de1_702_g;src=C;pri=4;
2R ProtHint intron 8446312 8446371 1 + . grp=43151_0:001cae_473_g;src=C;pri=4;
2R ProtHint intron 8011796 8011865 2 - . src=P;mult=1;pri=4;al_score=0.12;
2R ProtHint start 234524 234526 1 + . src=P;mult=1;pri=4;al_score=0.08;
The prediction of all hints with src=M will be enforced. Hints with src=C are 'chained evidence', ie they will only be incorporated if all members of the group (grp=...) can be incorporated in a single transcript. All other hints have src=P in the last column. Supported features in column 3 are intron , start , stop and CDSpart .
If RNA-Seq (and only RNA-Seq) data is provided to BRAKER as a bam-file, and if the genome is softmasked for repeats, BRAKER can automatically train UTR parameters for AUGUSTUS. After successful training of UTR parameters, BRAKER will automatically predict genes including coverage information form RNA-Seq data. Example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=file.bam --UTR=on
Warnings:
This feature is experimental!
--UTR=on is currently not compatible with bamToWig.py as released in AUGUSTUS 3.3.3; it requires the current development code version from the github repository (git clone https://github.com/Gaius-Augustus/Augustus.git).
--UTR=on increases memory consumption of AUGUSTUS. Carefully monitor jobs if your machine was close to maxing RAM without --UTR=on! Reducing the number of cores will also reduce RAM consumption.
UTR prediction sometimes improves coding sequence prediction accuracy, but not always. If you try this feature, carefully compare results with and without UTR parameters, afterwards (eg in UCSC Genome Browser).
For running BRAKER without UTR parameters, it is not very important whether RNA-Seq data was generated by a stranded protocol (because spliced alignments are 'artificially stranded' by checking the splice site pattern). However, for UTR training and prediction, stranded libraries may provide information that is valuable for BRAKER.
After alignment of the stranded RNA-Seq libraries, separate the resulting bam file entries into two files: one for plus strand mappings, one for minus strand mappings. Call BRAKER as follows:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam --stranded=+,-
--UTR=on
You may additionally include bam files from unstranded libraries. Those files will not used for generating UTR training examples, but they will be included in the final gene prediction step as unstranded coverage information, example call:
braker.pl --species=yourSpecies --genome=genome.fasta
--bam=plus.bam,minus.bam,unstranded.bam
--stranded=+,-,. --UTR=on
Warning: This feature is experimental and currently has low priority on our maintenance list!
The native mode for running BRAKER with RNA-Seq and protein data. This will call GeneMark-ETP, which will use RNA-Seq and protein hints for training GeneMark-ETP. Subsequently, AUGUSTUS is trained on 'high-confindent' genes (genes with very high extrinsic evidence support) from the GeneMark-ETP prediction and a set of genes is predicted by AUGUSTUS. In a last step, the predictions of AUGUSTUS and GeneMark-ETP are combined using TSEBRA.
Alignment of RNA-Seq reads
GeneMark-ETP utilizes Stringtie2 to assemble RNA-Seq data, which requires that the aligned reads (BAM files) contain the XS (strand) tag for spliced reads. Therefore, if you align your reads with HISAT2, you must enable the --dta option, or if you use STAR, you must use the --outSAMstrandField intronMotif option. TopHat alignments include this tag by default.
To call the pipeline in this mode, you have to provide it with a protein database using --prot_seq (as described in BRAKER with protein data), and RNA-Seq data either by their SRA ID so that they are downloaded by BRAKER, as unaligned reads in FASTQ format, and/or as aligned reads in bam format (as described in BRAKER with RNA-Seq data). You could also specify already processed extrinsic evidence using the --hints option. However, this is not recommend for a normal BRAKER run in ETPmode, as these hints won't be used in the GeneMark-ETP step. Only use --hints when you want to skip the GenMark-ETP step!
Examples of how you could run BRAKER in ETPmode:
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--rnaseq_sets_ids=SRA_ID1,SRA_ID2,SRA_ID3
braker.pl --genome=genome.fa --prot_seq=orthodb.fa
--bam=/path/to/SRA_ID1.bam,/path/to/SRA_ID2.bam
A preliminary protocol for integration of assembled subreads from PacBio ccs sequencing in combination with short read Illumina RNA-Seq and protein database is described at https://github.com/Gaius-Augustus/BRAKER/blob/master/docs/long_reads/long_read_protocol.md
We forked GeneMark-ETP and hard coded that StringTie will perform long read assembly in that particular version. If you want to use this 'fast-hack' version for BRAKER, you have to prepare the BAM file with long read to genome spliced alignments outside of BRAKER, eg:
T=48 # adapt to your number of threads
minimap2 -t${T} -ax splice:hq -uf genome.fa isoseq.fa > isoseq.sam
samtools view -bS --threads ${T} isoseq.sam -o isoseq.bam
Pull the adapted container:
singularity build braker3_lr.sif docker://teambraker/braker3:isoseq
Calling BRAKER3 with a BAM file of spliced-aligned IsoSeq Reads:
singularity exec -B ${PWD}:${PWD} braker3_lr.sif braker.pl --genome=genome.fa --prot_seq=protein_db.fa –-bam=isoseq.bam --threads=${T}
Warning Do NOT mix short read and long read data in this BRAKER/GeneMark-ETP variant!
Warning The accuracy of gene prediction here heavily depends on the depth of your isoseq data. We verified with PacBio HiFi reads from 2022 that given sufficient completeness of the assembled transcriptome you will reach similar results as with short reads. However, we also observed a drop in accuracy compared to short reads when using other long read data sets with higher error rates and less sequencing depth.
Please run braker.pl --help to obtain a full list of options.
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files: augustus.ab_initio.* . This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.
One or several command line arguments to be passed to AUGUSTUS, if several arguments are given, separate them by whitespace, ie "--first_arg=sth --second_arg=sth" . This may be be useful if you know that gene prediction in your particular species benefits from a particular AUGUSTUS argument during the prediction step.
Specifies the maximum number of threads that can be used during computation. BRAKER has to run some steps on a single thread, others can take advantage of multiple threads. If you use more than 8 threads, this will not speed up all parallelized steps, in particular, the time consuming optimize_augustus.pl will not use more than 8 threads. However, if you don't mind some threads being idle, using more than 8 threads will speed up other steps.
GeneMark-ETP option: run algorithm with branch point model. Use this option if you genome is a fungus.
Use the present config and parameter files if they exist for 'species'; will overwrite original parameters if BRAKER performs an AUGUSTUS training.
Execute CRF training for AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than HMM parameters. This increases runtime!
Change the parameter
Generate UTR training examples for AUGUSTUS from RNA-Seq coverage information, train AUGUSTUS UTR parameters and predict genes with AUGUSTUS and UTRs, including coverage information for RNA-Seq as evidence. This is an experimental feature!
If you performed a BRAKER run without --UTR=on, you can add UTR parameter training and gene prediction with UTR parameters (and only RNA-Seq hints) with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf
--threads=8 --skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify flaning_DNA value to the flanking region from the log file of your previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; modify somespecies to the species name used in your previous BRAKER run.
Add UTRs from RNA-Seq converage information to AUGUSTUS gene predictions using GUSHR. No training of UTR parameters and no gene prediction with UTR parameters is performed.
If you performed a BRAKER run without --addUTR=on, you can add UTRs results of a previous BRAKER run with the following command:
braker.pl --genome=../genome.fa --addUTR=on
--bam=../RNAseq.bam --workingdir=$wd
--AUGUSTUS_hints_preds=augustus.hints.gtf --threads=8
--skipAllTraining --species=somespecies
Modify augustus.hints.gtf to point to the AUGUSTUS predictions with hints from previous BRAKER run; modify some_new_working_directory to the location where BRAKER should store results of the additional BRAKER run; this run will not modify AUGUSTUS parameters. We recommend that you specify the original species of the original run with --species=somespecies . Otherwise, BRAKER will create an unneeded species parameters directory Sp_* .
If --UTR=on is enabled, strand-separated bam-files can be provided with --bam=plus.bam,minus.bam . In that case, --stranded=... should hold the strands of the bam files ( + for plus strand, - for minus strand, . for unstranded). Note that unstranded data will be used in the gene prediction step, only, if the parameter --stranded=... is set. This is an experimental feature! GUSHR currently does not take advantage of stranded data.
If --makehub and [email protected] (with your valid e-mail adress) are provided, a track data hub for visualizing results with the UCSC Genome Browser will be generated using MakeHub (https://github.com/Gaius-Augustus/MakeHub).
By default, GeneMark-ES/ET/EP/ETP uses a probability of 0.001 for predicting the donor splice site pattern GC (instead of GT). It may make sense to increase this value for species where this donor splice site is more common. For example, in the species Emiliania huxleyi , about 50% of donor splice sites have the pattern GC (https://media.nature.com/original/nature-assets/nature/journal/v499/n7457/extref/nature12221-s2.pdf, page 5).
Use a species-specific lineage, eg arthropoda_odb10 for an arthropod. BRAKER does not support auto-typing of the lineage.
Specifying a BUSCO-lineage invokes two changes in BRAKER R28 :
BRAKER will run compleasm with the specified lineage in genome mode and convert the detected BUSCO matches into hints for AUGUSTUS. This may increase the number of BUSCOs in the augustus.hints.gtf file slightly.
BRAKER will invoke best_by_compleasm.py to check whether the braker.gtf file that is by default generated by TSEBRA has the lowest amount of missing BUSCOs compared to the augustus.hints.gtf and the genemark.gtf file. If not, the following decision schema is applied to re-run TSEBRA to minimize the missing BUSCOs in the final output of BRAKER (always braker.gtf). If an alternative and better gene set is created, the original braker.gtf gene set is moved to a directory called braker_original. Information on what happened during the best_by_compleasm.py run is written to the file best_by_compleasm.log.
![best_by_busco[fig14]](https://images.downcodes.com/uploads/20250214/img_67aee79a11fd439.png)
Please note that using BUSCO to assess the quality of a gene set, in particular when comparing BRAKER to other pipelines, does not make sense once you specified a BUSCO lineage. We recommend that you use other measures to assess the quality of your gene set, eg by comparing it to a reference gene set or running OMArk.
BRAKER produces several important output files in the working directory.
braker.gtf: Final gene set of BRAKER. This file may contain different contents depending on how you called BRAKER
in ETPmode: Final gene set of BRAKER consisting of genes predicted by AUGUSTUS and GeneMark-ETP that were combined and filtered by TSEBRA.
otherwise: Union of augustus.hints.gtf and reliable GeneMark-ES/ET/EP predictions (genes fully supported by external evidence). In --esmode , this is the union of augustus.ab_initio.gtf and all GeneMark-ES genes. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present). This output is not necessarily better than augustus.hints.gtf, and it is not recommended to use it if BRAKER was run in ESmode.
braker.codingseq: Final gene set with coding sequences in FASTA format
braker.aa: Final gene set with protein sequences in FASTA format
braker.gff3: Final gene set in gff3 format (only produced if the flag --gff3 was specified to BRAKER.
Augustus/*: Augustus gene set(s) in as gtf/conding/aa files
GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+/ETP in GTF-format.
hintsfile.gff: The extrinsic evidence data extracted from RNAseq.bam and/or protein data.
braker_original/*: Genes predicted by BRAKER (TSEBRA merge) before compleasm was used to improve BUSCO completeness
bbc/*: output folder of best_by_compleasm.py script from TSEBRA that is used to improve BUSCO completeness in the final output of BRAKER
Output files may be present with the following name endings and formats:
Coding sequences in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
Protein sequence files in FASTA-format are produced if the flag --skipGetAnnoFromFasta was not set.
For details about gtf format, see http://www.sanger.ac.uk/Software/formats/GFF/. A GTF-format file contains one line per predicted exon. ตัวอย่าง:
HS04636 AUGUSTUS initial 966 1017 . + 0 transcript_id "g1.1"; gene_id "g1";
HS04636 AUGUSTUS internal 1818 1934 . + 2 transcript_id "g1.1"; gene_id "g1";
The columns (fields) contain:
seqname source feature start end score strand frame transcript ID and gene ID
If the --makehub option was used and MakeHub is available on your system, a hub directory beginning with the name hub_ will be created. Copy this directory to a publicly accessible web server. A file hub.txt resides in the directory. Provide the link to that file to the UCSC Genome Browser for visualizing results.
An incomplete example data set is contained in the directory BRAKER/example . In order to complete the data set, please download the RNA-Seq alignment file (134 MB) with wget :
cd BRAKER/example
wget http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam
In case you have trouble accessing that file, there's also a copy available from another server:
cd BRAKER/example
wget http://bioinf.uni-greifswald.de/augustus/datasets/RNAseq.bam
The example data set was not compiled in order to achieve optimal prediction accuracy, but in order to quickly test pipeline components. The small subset of the genome used in these test examples is not long enough for BRAKER training to work well.
Data corresponds to the last 1,000,000 nucleotides of Arabidopsis thaliana 's chromosome Chr5, split into 8 artificial contigs.
RNA-Seq alignments were obtained by VARUS.
The protein sequences are a subset of OrthoDB v10 plants proteins.
List of files:
genome.fa - genome file in fasta formatRNAseq.bam - RNA-Seq alignment file in bam format (this file is not a part of this repository, it must be downloaded separately from http://topaz.gatech.edu/GeneMark/Braker/RNAseq.bam)RNAseq.hints - RNA-Seq hints (can be used instead of RNAseq.bam as RNA-Seq input to BRAKER)proteins.fa - protein sequences in fasta formatThe below given commands assume that you configured all paths to tools by exporting bash variables or that you have the necessary tools in your $PATH.
The example data set also contains scripts tests/test*.sh that will execute below listed commands for testing BRAKER with the example data set. You find example results of AUGUSTUS and GeneMark-ES/ET/EP/ETP in the folder results/test* . Be aware that BRAKER contains several parts where random variables are used, ie results that you obtain when running the tests may not be exactly identical. To compare your test results with the reference ones, you can use the compare_intervals_exact.pl script as follows:
# Compare CDS features
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --verbose
# Compare transcripts
compare_intervals_exact.pl --f1 augustus.hints.gtf --f2 ../../results/test${N}/augustus.hints.gtf --trans --verbose
Several tests use --gm_max_intergenic 10000 option to make the test runs faster. It is not recommended to use this option in real BRAKER runs, the speed increase achieved by adjusting this option is negligible on full-sized genomes.
We give runtime estimations derived from computing on Intel(R) Xeon(R) CPU E5530 @ 2.40GHz .
The following command will run the pipeline according to Figure 3:
braker.pl --genome genome.fa --bam RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test1.sh , expected runtime is ~20 minutes.
The following command will run the pipeline according to Figure 4:
braker.pl --genome genome.fa --prot_seq proteins.fa --threads N --busco_lineage=lineage_odb10
This test is implemented in test2.sh , expected runtime is ~20 minutes.
The following command will run a pipeline that first trains GeneMark-ETP with protein and RNA-Seq hints and subsequently trains AUGUSTUS on the basis of GeneMark-ETP predictions. AUGUSTUS predictions are also performed with hints from both sources, see Figure 5.
Run with local RNA-Seq file:
braker.pl --genome genome.fa --prot_seq proteins.fa --bam ../RNAseq.bam --threads N --busco_lineage=lineage_odb10
This test is implemented in test3.sh , expected runtime is ~20 minutes.
Download RNA-Seq library from Sequence Read Archive (~1gb):
braker.pl --genome genome.fa --prot_seq proteins.fa --rnaseq_sets_ids ERR5767212 --threads N --busco_lineage=lineage_odb10
This test is implemented in test3_4.sh , expected runtime is ~35 minutes.
The training step of all pipelines can be skipped with the option --skipAllTraining . This means, only AUGUSTUS predictions will be performed, using pre-trained, already existing parameters. For example, you can predict genes with the command:
braker.pl --genome=genome.fa --bam RNAseq.bam --species=arabidopsis
--skipAllTraining --threads N
This test is implemented in test4.sh , expected runtime is ~1 minute.
The following command will run the pipeline with no extrinsic evidence:
braker.pl --genome=genome.fa --esmode --threads N
This test is implemented in test5.sh , expected runtime is ~20 minutes.
The following command will run BRAKER with training UTR parameters from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --UTR=on --threads N
This test is implemented in test6.sh , expected runtime is ~20 minutes.
The following command will add UTRs to augustus.hints.gtf from RNA-Seq coverage data:
braker.pl --genome genome.fa --bam RNAseq.bam --addUTR=on --threads N
This test is implemented in test7.sh , expected runtime is ~20 minutes.
There is currently no clean way to restart a failed BRAKER run (after solving some problem). However, it is possible to start a new BRAKER run based on results from a previous run -- given that the old run produced the required intermediate results. We will in the following refer to the old working directory with variable ${BRAKER_OLD} , and to the new BRAKER working directory with ${BRAKER_NEW} . The file what-to-cite.txt will always only refer to the software that was actually called by a particular run. You might have to combine the contents of ${BRAKER_NEW}/what-to-cite.txt with ${BRAKER_OLD}/what-to-cite.txt for preparing a publication. The following figure illustrates at which points BRAKER run may be intercepted.
![braker-intercept[fig8]](https://images.downcodes.com/uploads/20250214/img_67aee79a12cab310.png)
Figure 10: Points for intercepting a BRAKER run and reusing intermediate results in a new BRAKER run.
This option is only possible for BRAKER in ETmode or EPmode and ไม่ in ETPmode!
If you have access to an existing BRAKER output that contains hintsfiles that were generated from extrinsic data, such as RNA-Seq or protein sequences, you can recycle these hints files in a new BRAKER run. Also, hints from a separate ProtHint run can be directly used in BRAKER.
The hints can be given to BRAKER with --hints ${BRAKER_OLD}/hintsfile.gff option. This is illustrated in the test files test1_restart1.sh , test2_restart1.sh , test4_restart1.sh . The other modes (for which this test is missing) cannot be restarted in this way.
The GeneMark result can be given to BRAKER with --geneMarkGtf ${BRAKER_OLD}/GeneMark*/genemark.gtf option if BRAKER is run in ETmode or EPmode. This is illustrated in the test files test1_restart2.sh , test2_restart2.sh , test5_restart2.sh .
In ETPmode, you can either provide BRAKER with the results of the GeneMarkETP step manually, with --geneMarkGtf ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf , --traingenes ${BRAKER_OLD}/GeneMark-ETP/training.gtf , and --hints ${BRAKER_OLD}/hintsfile.gff (see test3_restart1.sh for an example), or you can specify the previous GeneMark-ETP results with the option --gmetp_results_dir ${BRAKER_OLD}/GeneMark-ETP/ so that BRAKER can search for the files automatically (see test3_restart2.sh for an example).
The trained species parameters for AGUSTUS can be passed with --skipAllTraining and --species $speciesName options. This is illustrated in test*_restart3.sh files. Note that in ETPmode you have to specify the GeneMark files as described in Option 2!
Before reporting bugs, please check that you are using the most recent versions of GeneMark-ES/ET/EP/ETP, AUGUSTUS and BRAKER. Also, check the list of Common problems, and the Issue list on GitHub before reporting bugs. We do monitor open issues on GitHub. Sometimes, we are unable to help you, immediately, but we try hard to solve your problems.
If you found a bug, please open an issue at https://github.com/Gaius-Augustus/BRAKER/issues (or contact [email protected] or [email protected]).
Information worth mentioning in your bug report:
Check in braker/yourSpecies/braker.log at which step braker.pl crashed.
There are a number of other files that might be of interest, depending on where in the pipeline the problem occurred. Some of the following files will not be present if they did not contain any errors.
braker/yourSpecies/errors/bam2hints.*.stderr - will give details on a bam2hints crash (step for converting bam file to intron gff file)
braker/yourSpecies/hintsfile.gff - is this file empty? If yes, something went wrong during hints generation - does this file contain hints from source “b2h” and of type “intron”? If not: GeneMark-ET will not be able to execute properly. Conversely, GeneMark-EP+ will not be able to execute correctly if hints from the source "ProtHint" are missing.
braker/yourSpecies/spaln/*err - errors reported by spaln
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP}.stderr - errors reported by GeneMark-ET/EP+/ETP
braker/yourSpecies/errors/GeneMark-{ET,EP,ETP).stdout - may give clues about the point at which errors in GeneMark-ET/EP+/ETP occured
braker/yourSpecies/GeneMark-{ET,EP,ETP}/genemark.gtf - is this file empty? If yes, something went wrong during executing GeneMark-ET/EP+/ETP
braker/yourSpecies/GeneMark-{ET,EP}/genemark.f.good.gtf - is this file empty? If yes, something went wrong during filtering GeneMark-ET/EP+ genes for training AUGUSTUS
braker/yourSpecies/genbank.good.gb - try a “grep -c LOCUS genbank.good.gb” to determine the number of training genes for training AUGUSTUS, should not be low
braker/yourSpecies/errors/firstetraining.stderr - contains errors from first iteration of training AUGUSTUS
braker/yourSpecies/errors/secondetraining.stderr - contains errors from second iteration of training AUGUSTUS
braker/yourSpecies/errors/optimize_augustus.stderr - contains errors optimize_augustus.pl (additional training set for AUGUSTUS)
braker/yourSpecies/errors/augustus*.stderr - contain AUGUSTUS execution errors
braker/yourSpecies/startAlign.stderr - if you provided a protein fasta file, something went wrong during protein alignment
braker/yourSpecies/startAlign.stdout - may give clues on at which point protein alignment went wrong
BRAKER complains that the RNA-Seq file does not correspond to the provided genome file, but I am sure the files correspond to each other!
Please check the headers of the genome FASTA file. If the headers are long and contain whitespaces, some RNA-Seq alignment tools will truncate sequence names in the BAM file. This leads to an error with BRAKER. Solution: shorten/simplify FASTA headers in the genome file before running the RNA-Seq alignment and BRAKER.
GeneMark fails!
(a) GeneMark by default only uses contigs longer than 50k for training. If you have a highly fragmented assembly, this might lead to "no data" for training. You can override the default minimal length by setting the BRAKER argument --min_contig=10000 .
(b) see "[something] failed to execute" below.
[something] failed to execute!
When providing paths to software to BRAKER, please use absolute, non-abbreviated paths. For example, BRAKER might have problems with --SAMTOOLS_PATH=./samtools/ or --SAMTOOLS_PATH=~/samtools/ . Please use SAMTOOLS_PATH=/full/absolute/path/to/samtools/ , instead. This applies to all path specifications as command line options to braker.pl . Relative paths and absolute paths will not pose problems if you export a bash variable, instead, or if you append the location of tools to your $PATH variable.
GeneMark-ETP in BRAKER dies with '/scratch/11232323': No such file or directory.
This appears to be related to sorting large files, and it's a system configuration depending problem. Solve it with export TMPDIR=/tmp/ before calling BRAKER via Singularity.
BRAKER cannot find the Augustus script XYZ...
Update Augustus from github with git clone https://github.com/Gaius-Augustus/Augustus.git . Do not use Augustus from other sources. BRAKER is highly dependent on an up-to-date Augustus. Augustus releases happen rather rarely, updates to the Augustus scripts folder occur rather frequently.
Does BRAKER depend on Python3?
It does. The python scripts employed by BRAKER are not compatible with Python2.
Why does BRAKER predict more genes than I expected?
If transposable elements (or similar) have not been masked appropriately, AUGUSTUS tends to predict those elements as protein coding genes. This can lead to a huge number genes. You can check whether this is the case for your project by BLASTing (or DIAMONDing) the predicted protein sequences against themselves (all vs. all) and counting how many of the proteins have a high number of high quality matches. You can use the output of this analysis to divide your gene set into two groups: the protein coding genes that you want to find and the repetitive elements that were additionally predicted.
I am running BRAKER in Anaconda and something fails...
Update AUGUSTUS and BRAKER from github with git clone https://github.com/Gaius-Augustus/Augustus.git and git clone https://github.com/Gaius-Augustus/BRAKER.git . The Anaconda installation is great, but it relies on releases of AUGUSTUS and BRAKER - which are often lagging behind. Please use the current GitHub code, instead.
Why and where is the GenomeThreader support gone?
BRAKER is a joint project between teams from University of Greifswald and Georgia Tech. While the group of Mark Bordovsky from Georgia Tech contributes GeneMark expertise, the group of Mario Stanke from University of Greifswald contributes AUGUSTUS expertise. Using GenomeThreader to build training genes for AUGUSTUS in BRAKER circumvents execution of GeneMark. Thus, the GenomeThreader mode is strictly speaking not part of the BRAKER project. The previous functionality of BRAKER with GenomeThreader has been moved to GALBA at https://github.com/Gaius-Augustus/GALBA. Note that GALBA has also undergone extension for using Miniprot instead of GenomeThreader.
My BRAKER gene set has too many BUSCO duplicates!
AUGUSTUS within BRAKER can predict alternative splicing isoforms. Also the merge of the AUGUSTUS and GeneMark gene set by TSEBRA within BRAKER may result in additional isoforms for a single gene. The BUSCO duplicates usually come from alternative splicing isoforms, ie they are expected.
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus , so you can simply do rm -rf ~/.augustus ; the folder might be residing in $PWD if your home directory was not writable).
I sit behind a firewall, compleasm cannot download the BUSCO files, what can I do? See Issue #785 (comment)
Since BRAKER is a pipeline that calls several Bioinformatics tools, publication of results obtained by BRAKER requires that not only BRAKER is cited, but also the tools that are called by BRAKER. BRAKER will output a file what-to-cite.txt in the BRAKER working directory, informing you about which exact sources apply to your run.
Always cite:
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013.
Stanke. M., Schöffmann, O., Morgenstern, B. and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62.
If you provided any kind of evidence for BRAKER, cite:
If you provided both short read RNA-Seq evidence and a large database of proteins, cite:
Gabriel, L., Bruna, T., Hoff, KJ, Ebel, M., Lomsadze, A., Borodovsky, M., Stanke, M. (2023). BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiV, doi: 10.1101/2023.06.10.54444910.1101/2023.01.01.474747.
Bruna, T., Lomsadze, A., Borodovsky, M. (2023). GeneMark-ETP: Automatic Gene Finding in Eukaryotic Genomes in Consistence with Extrinsic Data. bioRxiv, doi: 10.1101/2023.01.13.524024.
Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.
Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.
Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.
If the only source of evidence for BRAKER was a large database of protein sequences, cite:
If the only source of evidence for BRAKER was RNA-Seq data, cite:
Hoff, KJ, Lange, S., Lomsadze, A., Borodovsky, M. and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767-769.
Lomsadze, A., Paul DB, and Mark B. (2014) Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucleic Acids Research 42(15): e119--e119
If you called BRAKER3 with an IsoSeq BAM file, or if you envoked the --busco_lineage option, cite:
If you called BRAKER with the --busco_lineage option, in addition, cite:
Simão, FA, Waterhouse, RM, Ioannidis, P., Kriventseva, EV, & Zdobnov, EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19), 3210-3212.
Li, H. (2023). Protein-to-genome alignment with miniprot. Bioinformatics, 39(1), btad014.
Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.
If any kind of AUGUSTUS training was performed by BRAKER, check carefully whether you configured BRAKER to use NCBI BLAST or DIAMOND. One of them was used to filter out redundant training gene structures.
If you used NCBI BLAST, please cite:
Altschul, AF, Gish, W., Miller, W., Myers, EW and Lipman, DJ (1990). A basic local alignment search tool. J Mol Biol 215:403--410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, TL (2009). Blast+: architecture and applications. BMC bioinformatics, 10(1):421.
If you used DIAMOND, please cite:
If BRAKER was executed with a genome file and no extrinsic evidence, cite, then GeneMark-ES was used, cite:
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, YO and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome research, pages gr--081612, 2008.
Hoff, KJ, Lomsadze, A., Borodovsky, M. and Stanke, M. (2019). Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962:65-95, doi: 10.1007/978-1-4939-9173-0_5.
If BRAKER was run with proteins as source of evidence, please cite all tools that are used by the ProtHint pipeline to generate hints:
Bruna, T., Lomsadze, A., & Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026.
Buchfink, B., Xie, C., Huson, DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12:59-60.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, YO and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494--6506.
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research, 40(20), e161-e161.
Gotoh, O., Morita, M., Nelson, DR (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC bioinformatics, 15(1), 189.
If BRAKER was executed with RNA-Seq alignments in bam-format, then SAMtools was used, cite:
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16):2078-9.
Barnett, DW, Garrison, EK, Quinlan, AR, Strömberg, MP and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691-2
If BRAKER downloaded RNA-Seq libraries from SRA using their IDs, cite SRA, SRA toolkit, and HISAT2:
Leinonen, R., Sugawara, H., Shumway, M., & International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic acids research, 39(suppl_1), D19-D21.
SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.
If BRAKER was executed using RNA-Seq data in FASTQ format, cite HISAT2:
If BRAKER called MakeHub for creating a track data hub for visualization of BRAKER results with the UCSC Genome Browser, cite:
If BRAKER called GUSHR for generating UTRs, cite:
Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.
Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.
All source code, ie scripts/*.pl or scripts/*.py are under the Artistic License (see http://www.opensource.org/licenses/artistic-license.php).
[F1] EX = ES/ET/EP/ETP, all available for download under the name GeneMark-ES/ET/EP ↩
[F2] Please use the latest version from the master branch of AUGUSTUS distributed by the original developers, it is available from github at https://github.com/Gaius-Augustus/Augustus. Problems have been reported from users that tried to run BRAKER with AUGUSTUS releases maintained by third parties, ie Bioconda.
[F4] install with sudo apt-get install cpanminus ↩
[F6] The binary may eg reside in bamtools/build/src/toolkit ↩
[R0] Bruna, Tomas, Hoff, Katharina J., Lomsadze, Alexandre, Stanke, Mario, and Borodovsky, Mark. 2021. “BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database." NAR Genomics and Bioinformatics 3(1):lqaa108.↩
[R1] Hoff, Katharina J, Simone Lange, Alexandre Lomsadze, Mark Borodovsky, and Mario Stanke. 2015. “BRAKER1: Unsupervised Rna-Seq-Based Genome Annotation with Genemark-et and Augustus.” Bioinformatics 32 (5). Oxford University Press: 767--69.↩
[R2] Lomsadze, Alexandre, Paul D Burns, and Mark Borodovsky. 2014. “Integration of Mapped Rna-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm.” Nucleic Acids Research 42 (15). Oxford University Press: e119--e119.↩
[R3] Stanke, Mario, Mark Diekhans, Robert Baertsch, and David Haussler. 2008. “Using Native and Syntenically Mapped cDNA Alignments to Improve de Novo Gene Finding.” Bioinformatics 24 (5). Oxford University Press: 637--44.↩
[R4] Stanke, Mario, Oliver Schöffmann, Burkhard Morgenstern, and Stephan Waack. 2006. “Gene Prediction in Eukaryotes with a Generalized Hidden Markov Model That Uses Hints from External Sources.” BMC Bioinformatics 7 (1). BioMed Central: 62.↩
[R5] Barnett, Derek W, Erik K Garrison, Aaron R Quinlan, Michael P Strömberg, and Gabor T Marth. 2011. “BamTools: A C++ Api and Toolkit for Analyzing and Managing Bam Files.” Bioinformatics 27 (12). Oxford University Press: 1691--2.↩
[R6] Li, Heng, Handsaker, Bob, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. 2009. “The Sequence Alignment/Map Format and Samtools.” Bioinformatics 25 (16). Oxford University Press: 2078--9.↩
[R7] Gremme, G. 2013. “Computational Gene Structure Prediction.” PhD thesis, Universität Hamburg.↩
[R8] Gotoh, Osamu. 2008a. “A Space-Efficient and Accurate Method for Mapping and Aligning cDNA Sequences onto Genomic Sequence.” Nucleic Acids Research 36 (8). Oxford University Press: 2630--8.↩
[R9] Iwata, Hiroaki, and Osamu Gotoh. 2012. “Benchmarking Spliced Alignment Programs Including Spaln2, an Extended Version of Spaln That Incorporates Additional Species-Specific Features.” Nucleic Acids Research 40 (20). Oxford University Press: e161--e161.↩
[R10] Osamu Gotoh. 2008b. “Direct Mapping and Alignment of Protein Sequences onto Genomic Sequence.” Bioinformatics 24 (21). Oxford University Press: 2438--44.↩
[R11] Slater, Guy St C, and Ewan Birney. 2005. “Automated Generation of Heuristics for Biological Sequence Comparison.” BMC Bioinformatics 6(1). BioMed Central: 31.↩
[R12] Altschul, SF, W. Gish, W. Miller, EW Myers, and DJ Lipman. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215:403--10.↩
[R13] Camacho, Christiam, et al. 2009. “BLAST+: architecture and applications.“ BMC Bioinformatics 1(1): 421.↩
[R14] Lomsadze, A., V. Ter-Hovhannisyan, YO Chernoff, and M. Borodovsky. 2005. “Gene identification in novel eukaryotic genomes by self-training algorithm.” Nucleic Acids Research 33 (20): 6494--6506. doi:10.1093/nar/gki937.↩
[R15] Ter-Hovhannisyan, Vardges, Alexandre Lomsadze, Yury O Chernoff, and Mark Borodovsky. 2008. “Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training.” Genome Research . Cold Spring Harbor Lab, gr--081612.↩
[R16] Hoff, KJ 2019. MakeHub: Fully automated generation of UCSC Genome Browser Assembly Hubs. Genomics, Proteomics and Bioinformatics , in press, preprint on bioarXive, doi: https://doi.org/10.1101/550145.↩
[R17] Bruna, T., Lomsadze, A., & Borodovsky, M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics, 2(2), lqaa026. doi: https://doi.org/10.1093/nargab/lqaa026.↩
[R18] Kriventseva, EV, Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, FA, and Zdobnov, EM 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research, 47(D1), D807-D811.↩
[R19] Keilwagen, J., Hartung, F., Grau, J. (2019) GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962:161-177, doi: 10.1007/978-1-4939-9173-0_9.↩
[R20] Keilwagen, J., Wenk, M., Erickson, JL, Schattat, MH, Grau, J., Hartung F. (2016) Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89.↩
[R21] Keilwagen, J., Hartung, F., Paulini, M., Twardziok, SO, Grau, J. (2018) Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189.↩
[R22] SRA Toolkit Development Team (2020). SRA Toolkit. https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.[↩](#a22)
[R23] Kim, D., Paggi, JM, Park, C., Bennett, C., & Salzberg, SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology, 37(8):907-915.↩
[R24] Quinlan, AR (2014). BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics, 47(1):11-12.↩
[R25] Kovaka, S., Zimin, AV, Pertea, GM, Razaghi, R., Salzberg, SL, & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology, 20(1):1-13.↩
[R26] Pertea, G., & Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research, 9.↩
[R27] Huang, N., & Li, H. (2023). compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics, 39(10), btad595.↩
[R28] Bruna, T., Gabriel, L. & Hoff, KJ (2024). Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. arXiv, https://doi.org/10.48550/arXiv.2403.19416 .↩


