binnacle
1.0.0
ถึง
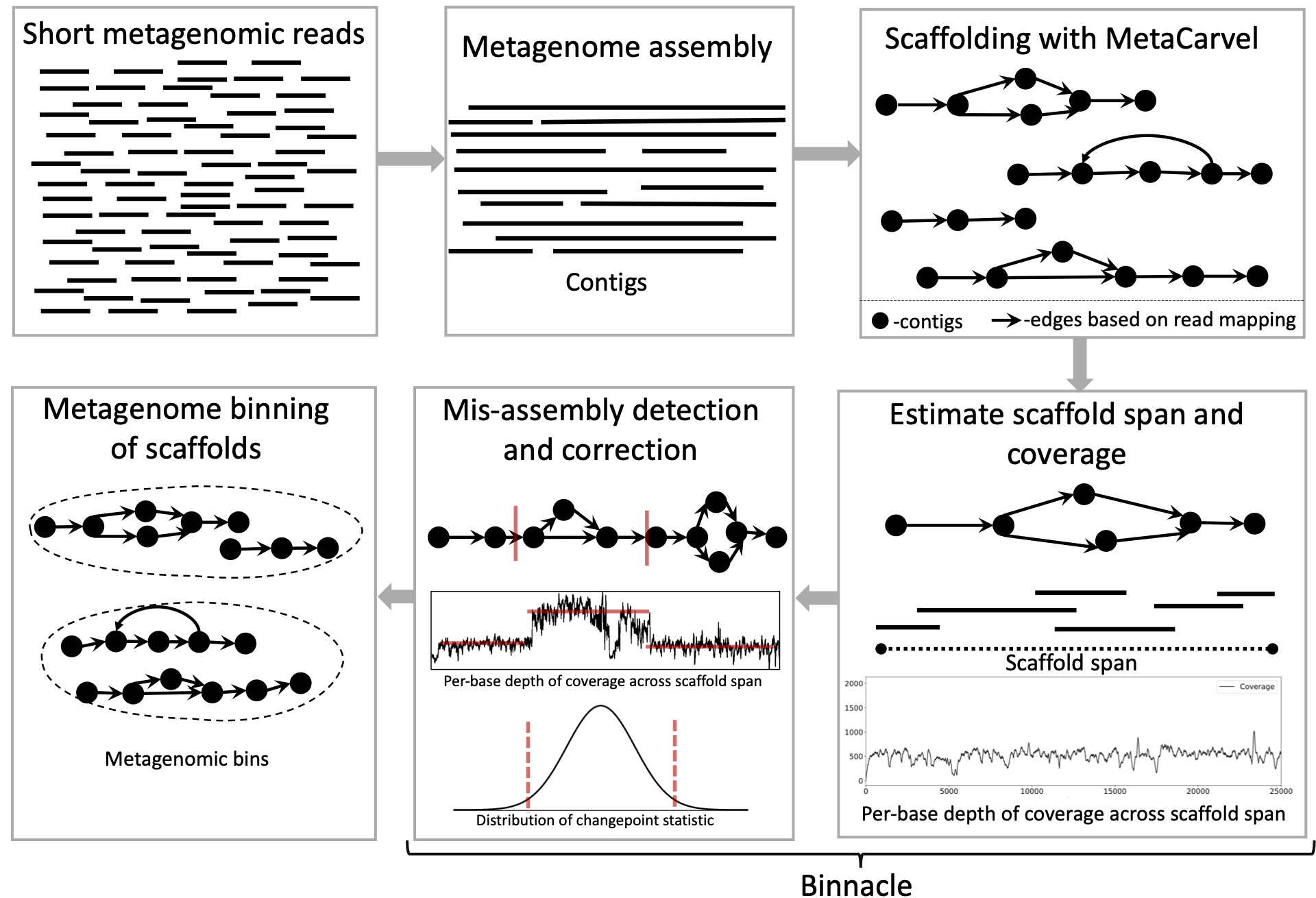
Binnacle คำนวณความครอบคลุมของโครงร่างกราฟอย่างแม่นยำและรวมเข้ากับวิธีการ binning ชั้นนำเช่น Metabat2, Maxbin 2.0 และ COCOCT การใช้กราฟนั่งร้านซึ่งตรงข้ามกับ contigs (วิธีการที่พบบ่อยที่สุด) สำหรับ binning ช่วยปรับปรุงความต่อเนื่องและคุณภาพของถังขยะ metagenomic และสามารถจับชุดองค์ประกอบอุปกรณ์เสริมที่กว้างขึ้นของจีโนมที่สร้างขึ้นใหม่
ในการเรียกใช้ Binnacle คุณจะต้องมี Python 3.7.x, Bedtools, Samtools, Biopython, NetworkX, Numpy และ Pandas
มีไฟล์ environment.yml และสามารถใช้เพื่อสร้างสภาพแวดล้อม conda ที่เหมาะสมในการเรียกใช้ binnacle เอกสารรายละเอียดเกี่ยวกับวิธีการติดตั้งแพ็คเกจเหล่านี้ได้รับที่นี่ เราใช้โครงร่างกราฟที่เป็นเอาต์พุตของเครื่องมือนั่งร้าน metacarvel ดังนั้นคุณจะต้องดาวน์โหลดและติดตั้ง metacarvel มีคู่มือการติดตั้งทีละขั้นตอนสำหรับ Metacarvel
โดยทั่วไปเมื่อคุณมีตัวอย่าง metagenomic หนึ่งหรือหลายตัวอย่างเราจำเป็นต้องรวบรวมนั่งร้านและถังเก็บ contigs/scaffolds จากแต่ละตัวอย่างเพื่อสร้างถังขยะ metagenomic เราแนะนำให้ใช้เมกาฮิทสำหรับการชุมนุมและ Metacarvel สำหรับการนั่งร้าน เราให้คำแนะนำผู้ช่วยในการทำงานผ่านการประกอบการนั่งร้านและขั้นตอนการประมาณค่าความครอบคลุมต่อฐานที่นี่
ทำตามขั้นตอนเหล่านี้เพื่อสร้างไฟล์สำหรับการเรียกใช้วิธีการ binning ด้วยกราฟนั่งร้าน:
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
โปรดชำระเงินวิกิสำหรับคำอธิบายโดยละเอียดเกี่ยวกับการตั้งค่าสภาพแวดล้อม Python วิธีการคำนวณความครอบคลุมและเวิร์กโฟลว์ทั่วไปเพื่อเรียกใช้ binnacle
ในการแสดงภาพโครงร่างกราฟเราขอแนะนำให้ใช้ Metagenomescope ซึ่งเป็นเบราว์เซอร์บนเว็บ อินพุตไปยัง metagenomescope คือ ASSEMBLY_GRAPH_FILTERED.GML เอกสารรายละเอียดเกี่ยวกับการติดตั้งและเรียกใช้ metagenomescope ได้รับที่นี่
โปรดอ้างอิง Muralidharan HS, Shah N, Meisel JS และ Pop M (2021) Binnacle: การใช้โครงนั่งร้านเพื่อปรับปรุงความต่อเนื่องและคุณภาพของถังขยะ metagenomic ด้านหน้า. microbiol 12: 638561 ดอย: 10.3389/fmicb.2021.638561
เครื่องมือนี้ยังอยู่ระหว่างการพัฒนา กรุณาเปิดปัญหาที่นี่บน GitHub หรือติดต่อเราหากคุณมีคำถามใด ๆ
Harihara Muralidharan: [email protected]
Nidhi Shah: [email protected]


