reference_database_creator
bug fix --in-silico-pcr --untrimmed
螃蟹( C评论右参考数据库一个复制子-乙阿塞德S equencing)是一种多功能软件程序,可以生成用于宏基因组分析的精选参考数据库。 CRABS 工作流程由七个模块组成:(i)从在线存储库下载数据; (ii) 将下载的数据导入CRABS格式; (iii) 通过计算机PCR 分析提取扩增子区域; (iv) 通过与计算机提取的条形码进行比对来检索没有引物结合区域的扩增子; (v) 通过多个过滤参数对本地数据库进行管理和子集化; (vi) 根据分类器要求以各种格式导出本地数据库; (vi) 后处理功能,即可视化,以探索并提供本地参考数据库的概要概述。这七个模块分为十八个功能,如下所述。此外,还为这十八个函数中的每一个函数提供了示例代码。最后,本 README 文档末尾提供了为 MiFish-E 引物集构建本地鲨鱼参考数据库的教程,以提供示例脚本供参考。
我们很高兴地宣布,CRABS 根据用户反馈进行了重大更新和代码重新设计,我们希望这将改善构建您自己的本地参考数据库的用户体验!
请在下面找到 CRABS v 1.0.0中添加的功能和改进列表:
现在可以通过克隆此 GitHub 存储库来手动下载 CRABS v 1.0.0 (有关详细信息,请参阅 4.1 手动安装)。我们将尽快更新Docker容器和conda包,以方便安装最新版本。
在您的研究项目中使用 CRABS 时,请引用以下论文:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS 是一个运行在典型 Unix/Linux 环境上的仅命令行工具包,并且专门用 python3 编写。然而,CRABS 利用 python 中的subprocess模块以 bash 语法运行多个命令,以规避 python 特定的特性并提高执行速度。我们提供了三种安装 CRABS 的方法。对于最新版本的 CRABS,我们建议通过克隆此 GitHub 存储库并分别安装 10 个依赖项进行手动安装(4.1 手动安装中提供了所有依赖项的安装说明)。 CRABS 也可以通过 Docker 和 conda 安装。这两种方法都可以通过自动共同安装所有依赖项来轻松安装。我们的目标是使 Docker 容器和 conda 包保持最新,尽管更新到最新版本可能会出现一定的延迟,尤其是 conda 包。以下是所有三种方法的详细信息。
对于手动安装,首先克隆 CRABS 存储库。此步骤要求 GitHub 可用于命令行(GitHub 的安装说明)。
git clone https://github.com/gjeunen/reference_database_creator.git
根据您的设置,CRABS 可能需要在您的系统上可执行。这可以使用下面的代码来实现。
chmod +x reference_database_creator/crabs
安装 CRABS 后,我们需要确保所有依赖项均已安装且可全局访问。最新版本的 CRABS(版本v 1.0.0 )在 Python 3.11.7(或 3.11.7 的任何兼容版本)上运行,并依赖于可能不是 Python 标准的五个 Python 模块以及五个外部软件程序。下面列出了所有依赖项,以及安装说明的链接。为每个模块和软件程序提供的版本号是 CRABS 开发的版本号。尽管也可以使用每个版本的兼容版本。
Python 模块:
外部软件程序:
安装 CRABS 和所有依赖项后,可以使用以下代码在整个操作系统中访问 CRABS。
export PATH="/path/to/crabs/folder:$PATH"
将/path/to/crabs/folder替换为操作系统上 GitHub 存储库文件夹的实际路径,即在上面的git clone命令期间创建的文件夹。将export代码添加到.bash_profile或.bashrc 文件中将使 CRABS 可以随时全局访问。
Docker 是一个开源项目,允许在与计算机隔离的“容器”内部署软件应用程序,并通过称为 Docker Engine 的虚拟主机操作系统运行。与虚拟机相比,运行 docker 的主要优点是它们使用的资源少得多。这种隔离意味着您可以在大多数操作系统上运行 Docker 容器,包括 Mac、Windows 和 Linux。您可能需要设置一个免费帐户才能使用 Docker Desktop。此链接很好地介绍了使用 Docker 的基础知识。以下链接可帮助您入门并了解 Docker 多元宇宙。
只需两个步骤即可让 Crabs 在您的计算机上运行。首先,在您的计算机上安装 Docker Desktop,这对大多数用户来说是免费的。以下是 Mac 版的说明;这是针对 Windows 计算机的说明,这是针对 Linux 计算机的说明(支持大多数主要 Linux 平台)。一旦安装并运行了 Docker Desktop(桌面应用程序必须运行,您才能在命令行上使用任何 docker 命令),您只需“拉取”我们的 Crabs 镜像,就可以开始了:
docker pull quay.io/swordfish/crabs:0.1.7
虽然 Docker 应用程序的安装很容易,但一开始使用这些应用程序可能会有点棘手。为了帮助您入门,我们提供了一些使用 Docker 版本的螃蟹的示例命令。这些示例可以在此存储库的 docker_intro 文件夹中找到。通过这些示例,您应该能够完成整个参考数据库的设置并做好准备。我们将继续扩展这些示例,并在许多不同的情况下进行测试。请在“问题”选项卡中提出问题并提供反馈。
要安装 conda 软件包,必须先安装 conda。请参阅此链接了解详细信息。如果已安装 conda,则最好在安装 CRABS 之前使用conda update conda更新 conda 工具。
安装 conda 后,请按照以下步骤安装 CRABS 和所有依赖项。确保按照下面显示的顺序输入命令。
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
输入安装命令后,conda 将处理请求(这可能需要一分钟左右),然后显示将安装的所有软件包和程序,并要求确认。输入y开始安装。完成后,CRABS 应该准备就绪。
我们已在 Mac 和 Linux 系统上测试了此安装。我们尚未在适用于 Linux 的 Windows 子系统 (WSL) 上进行测试。
使用下面的代码检查CRABS是否安装成功并拉取帮助信息。
crabs -h帮助信息将十八个函数分为不同的组,每个组在顶部列出该函数,在下面列出必需和可选的参数。

CRABS包含七个模块,包含十八个功能:
模块 1:从在线存储库下载数据
--download-taxonomy : 下载 NCBI 分类信息;--download-bold :从生命条形码数据库(BOLD)下载序列数据;--download-embl :从欧洲核苷酸档案库(ENA;EMBL)下载序列数据;--download-mitofish : 从MitoFish数据库下载序列数据;--download-ncbi :从国家生物技术信息中心(NCBI)下载序列数据。模块2:将下载的数据导入CRABS格式
--import :将下载的序列或自定义条形码导入为 CRABS 格式;--merge :将不同的 CRABS 格式的文件合并到一个文件中。模块 3:通过计算机PCR 分析提取扩增子区域
--in-silico-pcr :通过定位和删除引物结合区域从下载的数据中提取扩增子。模块 4:检索没有引物结合区域的扩增子
--pairwise-global-alignment :通过将下载的序列与计算机提取的条形码对齐来检索没有引物结合区域的扩增子。模块 5:通过多个过滤参数对本地数据库进行管理和子集化
--dereplicate :丢弃重复序列;--filter : 通过多个过滤参数丢弃序列;--subset :对本地数据库进行子集化以保留或排除指定的分类组。模块六:导出本地数据库
--export :根据要使用的分类分类器的要求将 CRABS 格式的数据库导出为各种格式。模块 7:后处理功能,探索并提供本地参考数据库的摘要概述
--diversity-figure :创建水平条形图,显示参考数据库中包含的每个指定级别的物种和序列组的数量;--amplicon-length-figure :创建一个折线图,描绘按分类组分隔的扩增子长度分布;--phylogenetic-tree :使用目标物种列表的参考数据库中的条形码创建系统发育树;--amplification-efficiency-figure :创建一个条形图,显示引物结合区域中的不匹配情况;--completeness-table :创建包含分类组条形码可用性的电子表格。CRABS 可以从四个在线存储库下载初始测序数据,包括 (i) BOLD、(ii) EMBL、(iii) MitoFish 和 NCBI。从版本v 1.0.0开始,从每个存储库下载数据都分为自己的功能。另外,CRABS在下载数据后不会自动格式化数据,以增加灵活性,并在数据下载失败时进行调试。
除了下载序列数据之外,CRABS 还能够下载 NCBI 分类信息,CRABS 使用该信息为每个序列创建分类谱系。
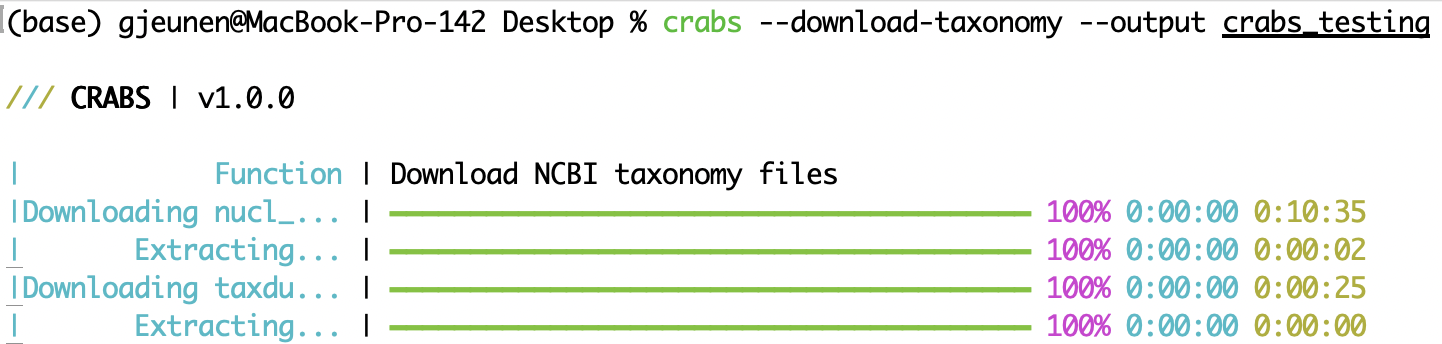
--download-taxonomy要为参考数据库中每个下载的序列分配分类谱系(参见 5.2 模块 2),需要下载分类信息。 CRABS 利用 NCBI 的分类法并将三个特定文件下载到您的计算机:(i) 将登录号链接到分类 ID 的文件 ( nucl_gb.accession2taxid ),(ii) 包含有关与每个分类 ID 关联的系统发育名称的信息的文件 ( names.dmp ),以及 (iii) 包含分类 ID 如何链接信息的文件 ( nodes.dmp )。可以使用--output参数指定下载文件的输出目录。要排除文件nucl_gb.accession2taxid或文件names.dmp和nodes.dmp ,可以分别提供--exclude acc2tax或--exclude taxdump参数。下面的第一个代码不会下载任何文件,因为acc2tax和taxdump都是为--exclude参数提供的。第二行代码将所有三个文件下载到子目录--output crabs_testing 。下面的屏幕截图显示了执行这行代码时打印到控制台的内容。
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold BOLD 序列通过 BOLD 网站下载。输出文件的结构为两行 fasta 文档,可以使用--output参数指定。用户可以使用--taxon参数指定要下载的分类组。当用户想要下载多个分类组时,我们建议编写一个简单的 for 循环(下面提供的示例),从而限制每个实例从 BOLD 下载的数据量。但是,如果仅对有限数量的分类组感兴趣,则分类组名称也可以用|分隔。 (下面提供的示例)。我们还建议用户检查要下载的分类组名称是否在粗体存档中列出,或者是否需要使用替代名称。例如,指定--taxon Chondrichthyes将不会从 BOLD 下载所有软骨鱼序列,因为该类名称未在 BOLD 中列出。在这种情况下,用户应该使用--taxon Elasmobranchii 。用户还可以通过提供--marker参数来指定将下载限制为特定的遗传标记。当对多个遗传标记感兴趣时,标记名称应以|分隔。 。 BOLD 上的四个主要 DNA 条形码标记是COI-5P 、 ITS 、 matK和rbcL 。 --marker参数的输入区分大小写。
推荐方法:一个简单的 for 循环,用于从 BOLD 下载多个分类组的数据(推荐方法)。下面的代码首先下载板鳃亚纲的数据,然后下载分配给哺乳动物的序列。下载的数据将写入子目录--output crabs_testing并放置在两个单独的文件中,指示哪些数据属于哪个分类组,即crabs_testing/bold_Elasmobranchii.fasta和crabs_testing/bold_Mammalia.fasta 。
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

替代选项:除了推荐的 for 循环之外,还可以通过使用|分隔名称来一次提供多个分类单元名称。 。
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl EMBL 的序列可通过 ENA FTP 站点下载。 EMBL 文件将首先以“.fasta.gz”格式下载,下载完成后会自动解压缩。与 BOLD 或 NCBI 相比,该数据库在选择性下载方面没有提供足够的灵活性。相反,EMBL 数据分为 15 个税务部门,可以单独下载。可以使用--taxon参数指定要下载的税务部门。由于每个税务部门都分为多个文件,因此在名称后提供*以下载所有文件。用户还可以通过写下完整的文件名来下载特定文件。下面提供了所有 15 个税务划分选项的列表。可以使用--output参数指定输出目录和文件名。
税务部门列表:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS还可以下载MitoFish数据库。该数据库是一个两行的 fasta 文件。可以使用--output参数指定输出目录和文件名。
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi NCBI 数据库中的序列通过 Entrez 编程实用程序下载。 NCBI 允许从各种数据库下载数据,用户可以使用--database参数指定。对于大多数用户来说, --database nucleotide数据库最适合构建本地参考数据库。
要指定要从 NCBI 下载的数据,用户可以通过--query参数提供搜索。精心设计 NCBI 检索可能很困难。构建搜索查询的一个好方法是使用 NCBI 网页搜索窗口。从此链接中,首先进行初始搜索并按 Enter 键。这将带您进入结果页面,您可以在其中进一步优化搜索。在下面的屏幕截图中,我们通过将序列长度限制在 100 - 25,000 bp 之间并且仅包含线粒体序列来进一步细化搜索。用户可以将文本复制粘贴到网站上的“搜索详细信息”框中,并将其放在引号中提供给--query参数。使用 NCBI 网页搜索窗口的另一个好处是,网页将显示有多少序列与您的搜索查询匹配,这应该与 CRABS 报告的序列数量相匹配。此网页提供了有关使用 NCBI 网页上的搜索功能的进一步简短教程,该教程是我们团队为额外信息而编写的。
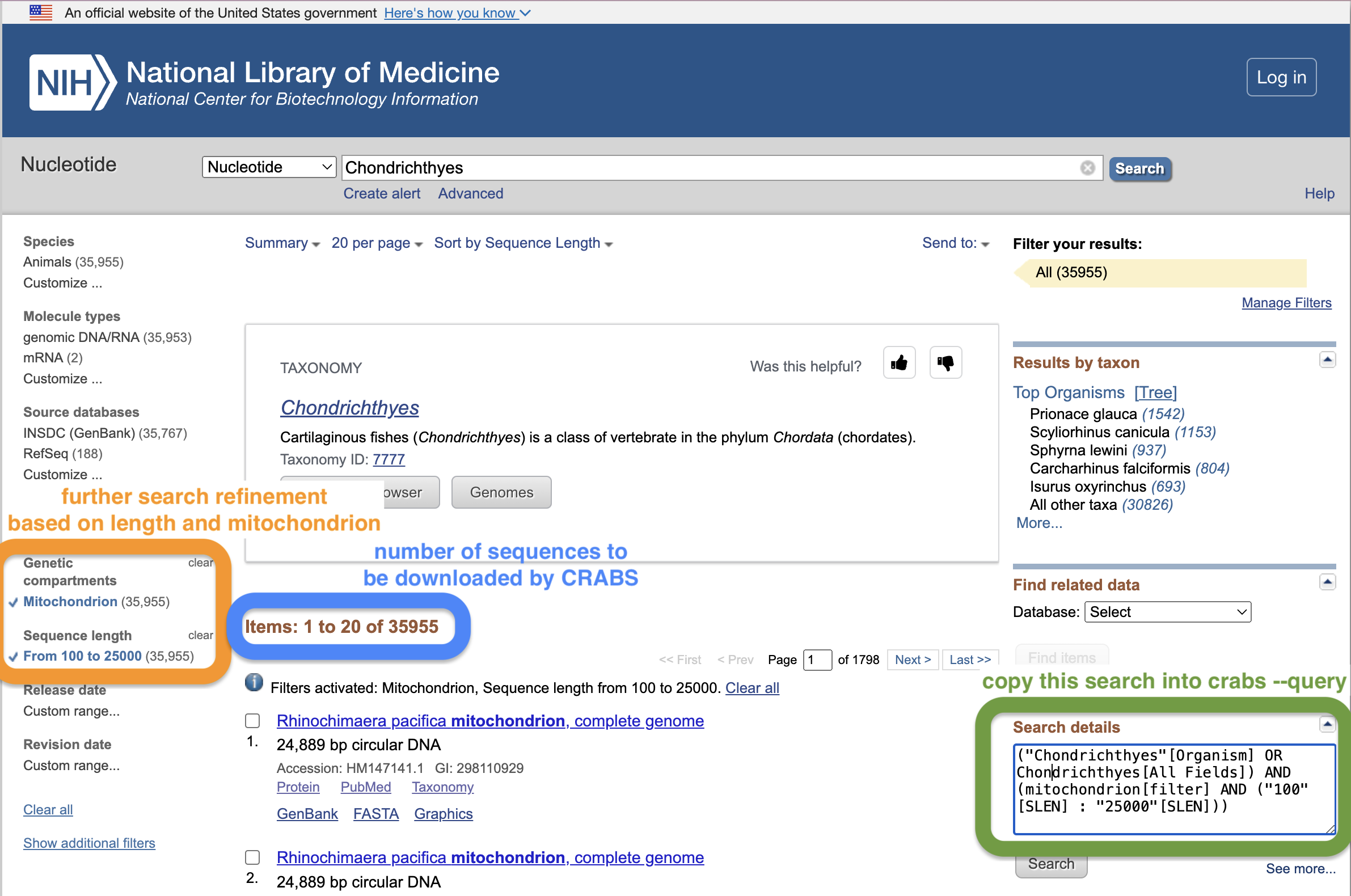
除了搜索查询 ( --query ) 之外,用户还可以使用--species参数下载物种列表的序列数据,进一步限制搜索项。 --species参数要么采用由+分隔的物种名称输入字符串,要么采用文档中每行一个物种名称的输入 .txt 文件。 --batchsize参数为用户提供从 NCBI 网站批量下载 N 序列的选项。该参数默认为 5,000。不建议将此值增加到 5,000 以上,因为如果一次下载太多序列,NCBI 服务器很可能会断开下载。 --email参数允许用户指定其电子邮件地址,这是访问 NCBI 服务器所必需的。最后,可以使用--output参数指定输出目录和文件名。
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import下载在线存储库中的数据后,需要使用--import函数将文件导入到 CRABS 中。 CRABS 格式构成每个序列的单个制表符分隔行,包含所有信息,包括 (i) 序列 ID、(ii) 从初始下载解析的分类名称、(iii) NCBI 分类单元 ID 号、(iv) 根据 NCBI 分类的分类谱系,和(v)序列。 CRABS 将尝试获取每个序列的 NCBI 登录号作为序列 ID。如果序列不包含登录号,即未保藏在 NCBI 上,CRABS 将使用以下格式生成唯一的序列 ID: crabs_*[num]*_taxonomic_name 。输入文档的格式使用--import-format参数指定,并指定从中下载数据的存储库的名称,即BOLD 、 EMBL 、 MITOFISH或NCBI 。 CRABS 创建的分类谱系基于 NCBI 分类,并且 CRABS 需要使用--download-taxonomy函数下载的三个文件,即--names 、 --nodes和--acc2tax 。从版本v 1.0.0开始,CRABS 能够解析同义词和不可接受的名称,以将大量序列和多样性纳入本地参考数据库中。可以使用--ranks参数指定要包含在分类谱系中的分类等级。虽然可以包含任何分类排名,但我们建议使用以下输入来包含大多数分类分类器的所有必要信息--ranks 'superkingdom;phylum;class;order;family;genus;species' 。输出文件可以使用--output参数指定,并且是一个简单的 .txt 文件。在终端窗口中,CRABS 打印导入序列数量的结果,以及无法生成分类谱系的任何序列。
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

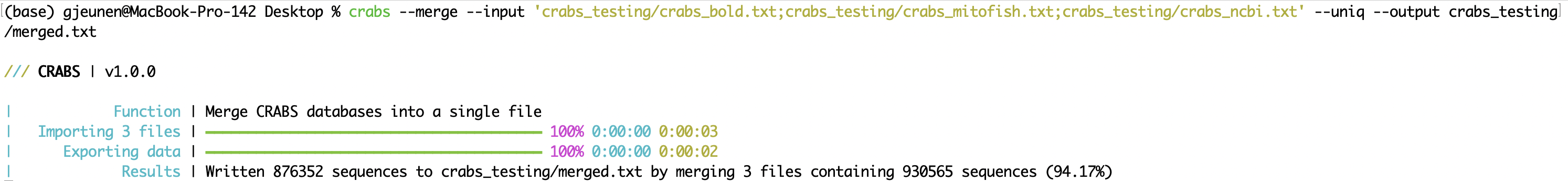
--merge当从多个在线存储库下载序列数据时,可以使用--merge函数在导入后将文件合并为单个文件(请参阅 5.2.1 --import )。可以使用--input参数输入要合并的输入文件,文件之间用; 。一个序列在存放到各个在线存储库时可能会被多次下载。使用--uniq参数仅保留每个入藏号的单个版本。可以使用--output参数指定输出文件。在终端窗口中,CRABS 打印合并序列数的结果,以及使用--uniq参数时保留的序列数。
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
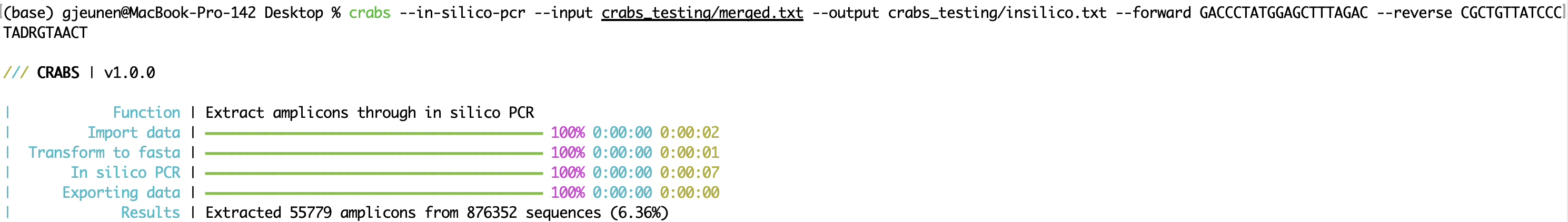
CRABS 通过进行计算机PCR(功能: --in-silico-pcr )提取引物组的扩增子区域。 CRABS 使用 cutadapt v 4.4进行计算机PCR,以提高传统 Python 代码的执行速度。可以分别使用“ --input ”和“ --output ”参数指定输入和输出文件名。正向和反向引物均应分别使用“ --forward ”和“ --reverse ”参数在 5'-3' 方向提供。 CRABS 将反向互补反向引物。从版本v 1.0.0开始,CRABS 能够使用单个计算机PCR 分析在两个方向上保留条形码。因此,无需进行反向互补步骤和重新运行计算机PCR,从而显着提高执行速度。要保留找不到引物结合区域的序列,可以为--untrimmed参数指定输出文件。可以使用--mismatch参数指定引物结合区域中发现的最大允许错配数,默认设置为 4。最后,计算机PCR 分析可以在 CRABS 中进行多线程。默认情况下使用最大线程数,但用户可以使用--threads参数指定要使用的线程数。
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
当参考序列存入在线数据库时,通常的做法是从参考序列中删除引物结合区域。因此,当使用与--in-silico-pcr函数中搜索的相同正向和/或反向引物生成参考序列时, --in-silico-pcr函数将无法恢复该序列的扩增子区域。参考序列。为了解决这种可能性,CRABS 可以选择运行使用 VSEARCH v 2.16.0实现的成对全局比对,以提取参考序列不包含完整正向和反向引物结合区域的扩增子区域。为了实现这一点, --pairwise-global-alignment函数使用--input参数接收最初下载的数据库文件。要搜索的数据库是--in-silico-pcr的输出文件,可以使用--amplicons参数指定。可以使用--output参数指定输出文件。引物序列仅用于计算碱基对长度,可以使用--forward和--reverse参数进行设置。由于--pairwise-global-alignment函数对于大型数据库可能需要很长时间才能运行,因此可以使用--size-select参数限制序列长度以加快该过程。可以分别使用--percent-identity和--coverage参数指定最小百分比身份和查询覆盖率。 --percent-identity应以 0 到 1 之间的百分比值形式提供(例如,95% = 0.95),而--coverage应以 0 到 100 之间的百分比值形式提供(例如,95% = 95)。默认情况下, --pairwise-global-alignment函数仅限于保留引物序列未完全存在于参考序列中的序列(比对在正向或反向引物的长度内开始或结束)。当提供--all-start-positions参数时,当发现比对超出引物结合区域范围时,将包含正命中(由于--in-silico-pcr函数由于引物结合区域中有太多不匹配而错过)引物结合区)。我们不建议使用--all-start-positions ,因为当引物中存在超过 4 个不匹配时,使用--in-silico-pcr函数的指定引物组扩增条形码的可能性很小 -结合区域。
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
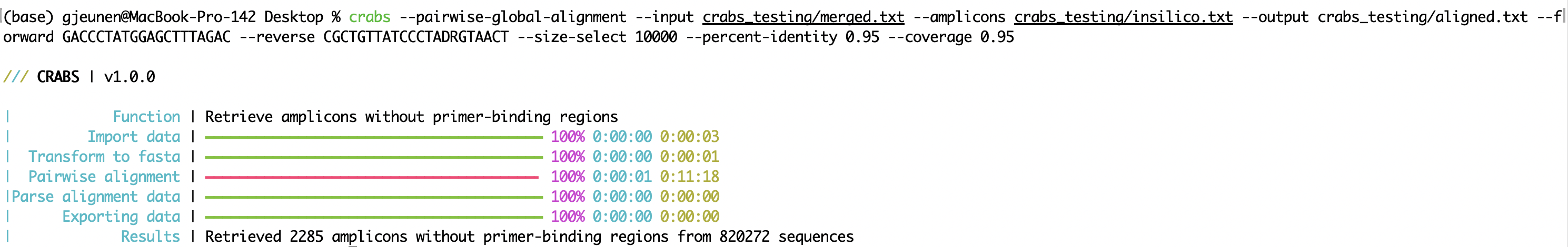
--pairwise-global-alignment的代码执行速度当 CRABS 处理大型序列文件时,即使支持多线程, --pairwise-global-alignment函数也可能需要花费大量时间来执行。自从更新到 CRABS v 1.0.0以来,从--import到--export都有相同的文件结构,从而使函数能够以任何顺序执行。虽然我们仍然建议遵循 CRABS 工作流程的顺序,但在--in-silico-pcr函数之前执行--dereplicate和--filter函数时,可以显着加快--pairwise-global-alignment函数的速度。通过在--in-silico-pcr之前执行这些管理步骤,CRABS 为--pairwise-global-alignment函数处理所需的序列数量将显着减少。
注意 1 :在--in-silico-pcr之前执行--filter函数时,请确保省略任何直接影响序列的参数,因为--filter会基于整个序列而不是提取的扩增子。因此,省略以下参数: --minimum-length 、 --maximum-length 、 --maximum-n 。
注 2 :在--in-silico-pcr之前执行--dereplicate和--filter函数时,建议在--pairwise-global-alignment之后再次运行这两个函数,因为现在可以进一步整理数据库扩增子被提取。
一旦通过--in-silico-pcr和--pairwise-global-alignment函数提取了引物集的所有潜在条形码,本地参考数据库就可以使用各种函数(包括--dereplicate在 CRABS 中进行进一步的整理和子集化、 --filter和--subset 。
--dereplicate第一种管理方法是使用--dereplicate函数取消复制本地参考数据库。对于某些分类单元,此时本地参考数据库中可能包含多个相同的条形码。当不同的研究小组沉积了相同的序列,或者如果提取的条形码中不包含分类单元序列之间的特异性内变异,则可能会发生这种情况。最好删除这些相同的参考条形码以加速分类分配,并改进分类分配结果(特别是对于提供有限数量结果的分类分类器,即BLAST)。
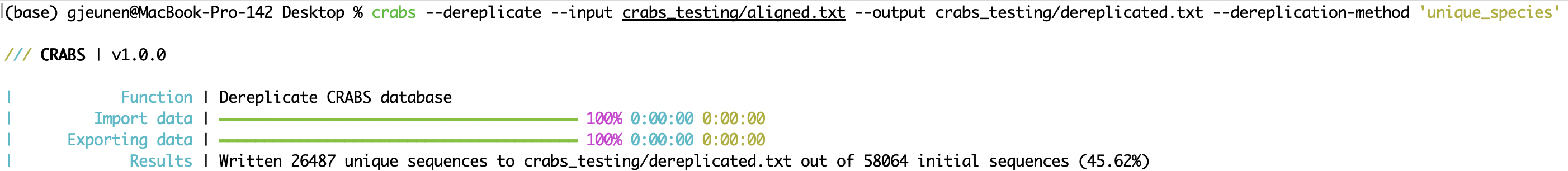
可以分别使用--input和--output参数指定输入和输出文件。 CRABS提供了三种去复制方法,可以使用--dereplication-method参数指定,包括:
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
--filter第二种管理方法是使用--filter函数使用各种参数来过滤本地参考数据库。可以分别使用--input和--output参数指定输入和输出文件。从版本v 1.0.0开始。 CRABS 结合了基于六个参数的过滤,包括:
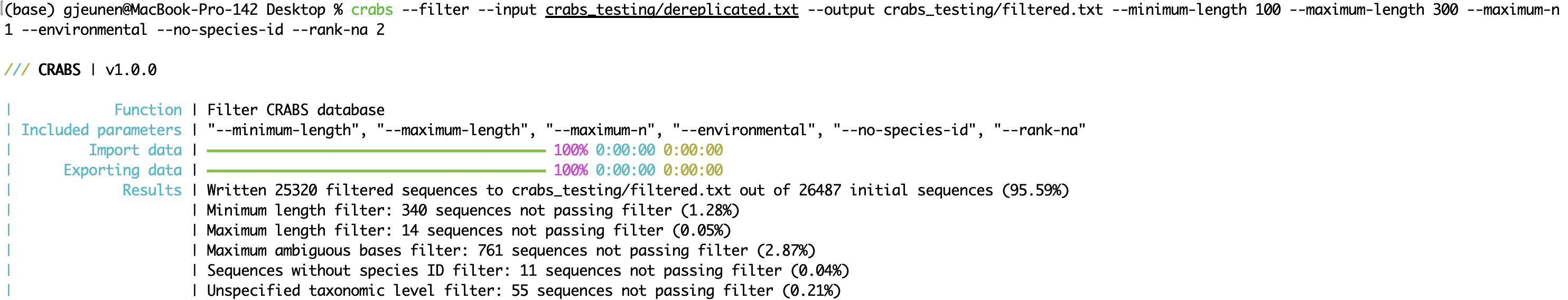
--minimum-length :数据库中保留的扩增子的最小序列长度;--maximum-length :数据库中保留的扩增子的最大序列长度;--maximum-n : 丢弃具有 N 个或更多不明确碱基 ( N ) 的扩增子;--environmental :从数据库中丢弃环境序列;--no-species-id :丢弃没有物种名称的序列;--rank-na :丢弃具有 N 个或更多未指定分类级别的序列。 crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
--subset CRABS 中包含的第三种也是最后一种管理方法是使用--subset函数对本地参考数据库进行子集化,以包含(参数: --include )或排除(参数: --exclude )特定分类单元。此功能允许从与研究问题不相关的分类组中删除参考条形码。由于引物组潜在的非特异性扩增,这些分类组可能已被纳入当地参考数据库。 --subset的另一个用例是删除已知的错误序列。
对于基于机器学习 (IDTAXA) 或 k-mer 距离 (SINTAX) 的分类学分类器,通过仅包含已知在采样区域中出现的类群并排除未知的密切相关物种来对参考数据库进行子集化可能是有益的发生在该区域以增加这些分类器获得的分类分辨率并获得改进的分类分配结果。
可以分别使用--input和--output参数指定输入和输出文件。 --include和--exclude参数可以接受由 ; 分隔的分类群列表;或每行包含一个分类单元名称的 .txt 文件。
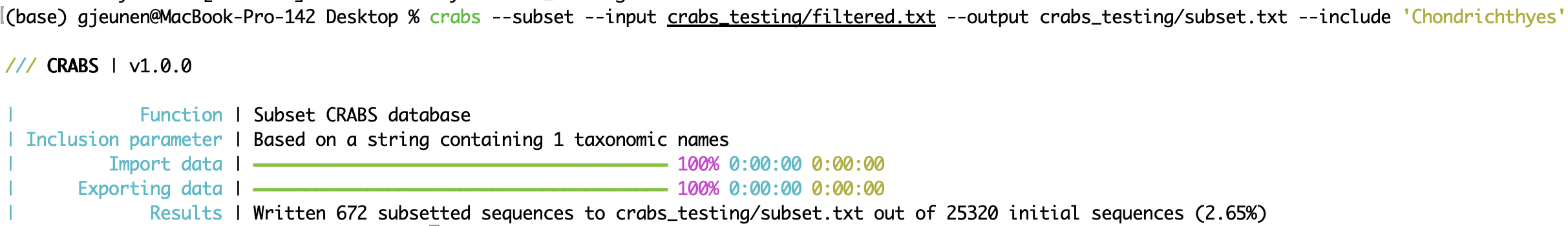
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
参考数据库最终确定后,可以将其导出为各种格式,以适应大多数将分类法分配给宏基因组数据的软件工具所需的规范。可以分别使用--input和--output参数指定输入和输出文件。从版本v 1.0.0开始,CRABS 合并了六种不同分类器的参考数据库格式(参数: --export-format ),包括:
--export-format 'sintax' : SINTAX 分类器合并到 USEARCH 和 VSEARCH 中;--export-format 'rdp' :RDP 分类器是一个广泛用于微生物组研究的独立程序;--export-format 'qiime-fasta'和--export-format 'qiime-text' :可用于在 QIIME 和 QIIME2 中分配分类 ID;--export-format 'dada2-species'和--export-format 'dada2-taxonomy' :可用于在 DADA2 中分配分类 ID;--export-format 'idt-fasta'和--export-format 'idt-text' :IDTAXA 分类器是包含在 DECIPHER R 包中的机器学习算法;--export-format 'blast-notax' :为BlastN和Megablast创建一个本地BLAST参考数据库,其中输出不提供分类ID,而是列出登录号;--export-format 'blast-tax' :为BlastN和Megablast创建本地BLAST参考数据库,其中输出提供分类学ID和登录号。 crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

虽然将本地参考数据库导出到单个格式(除了分类器除了在多个文件上拆分参考数据库的分类器,即大多数用户都足够如果用户想比较不同的分类分类器之间的结果,请参考多种格式。下面提供了一个示例,以在Sintax,RDP和IDTAXA格式中导出本地参考数据库。
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

一旦完成参考数据库,CRAB可以运行五个后处理功能来探索并提供概述局部参考数据库的概述,包括(i) --diversity-figure ,(ii) - (ii) --amplicon-length-figure ,((( iii) --phylogenetic-tree ,(iv) --amplification-efficiency-figure和(v) --completeness-table 。
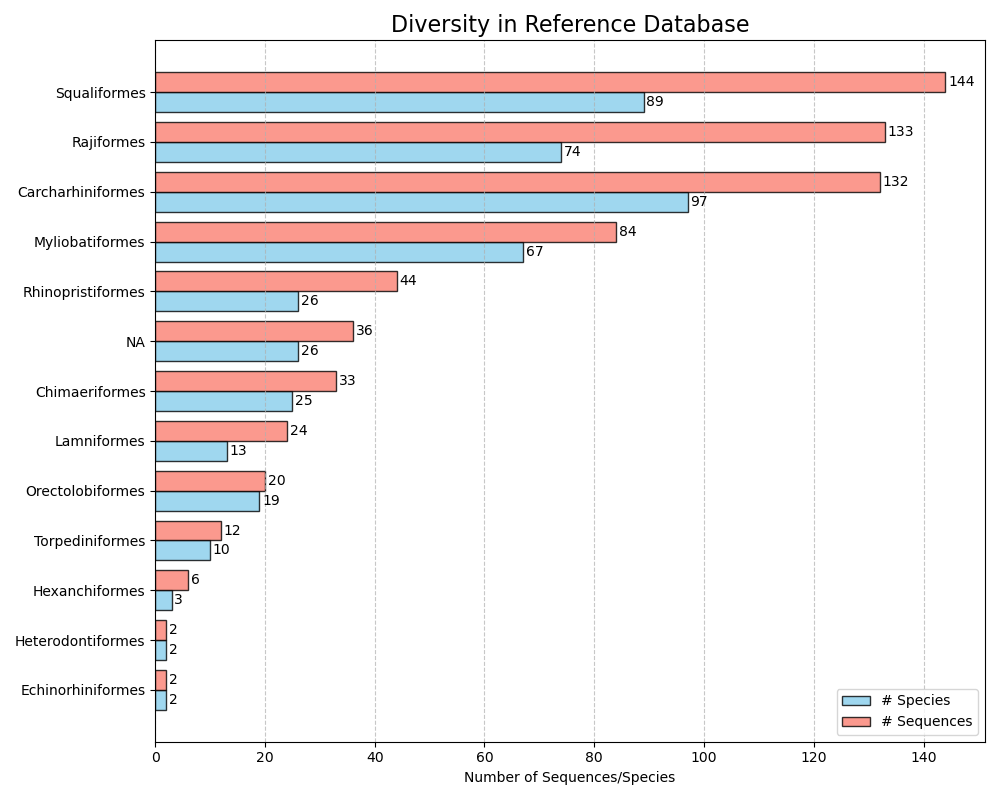
--diversity-figure --diversity-figure函数可为参考数据库中的每个分类群组提供一个水平棒图(蓝色)数量(蓝色)和每个分类学组的序列数(橙色)。用户可以指定分类等级,以将参考数据库用--tax-level参数拆分。税收水平是--import功能中它出现的等级的数量。例如,如果--ranks 'superkingdom;phylum;class;order;family;genus;species'基于超级界的--import拆分,则需要--tax-level 1 ,门= - 塔克斯级= --tax-level 2 , class = --tax-level 3等。可以使用--input参数指定CRABS格式的输入文件。以.png格式的图将写入输出文件,可以使用--output参数指定。
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
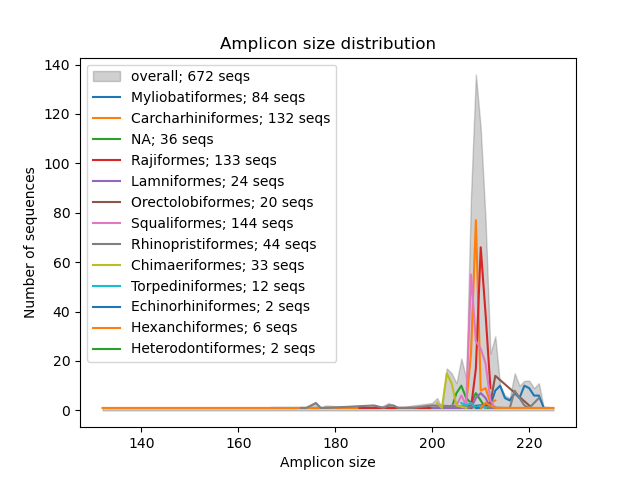
--amplicon-length-figure --amplicon-length-figure函数产生的线图显示了扩增子长度的范围。参考数据库中所有序列的扩增子长度的总范围均以阴影灰色显示,而每个分类学组的结果拆分(参数: --tax-level )被彩色线叠加。此外,该图例显示分配给每个分类组的序列数量以及参考数据库中的序列总数。可以使用--input参数指定CRABS格式的输入文件。以.png格式的图将写入输出文件,可以使用--output参数指定。
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
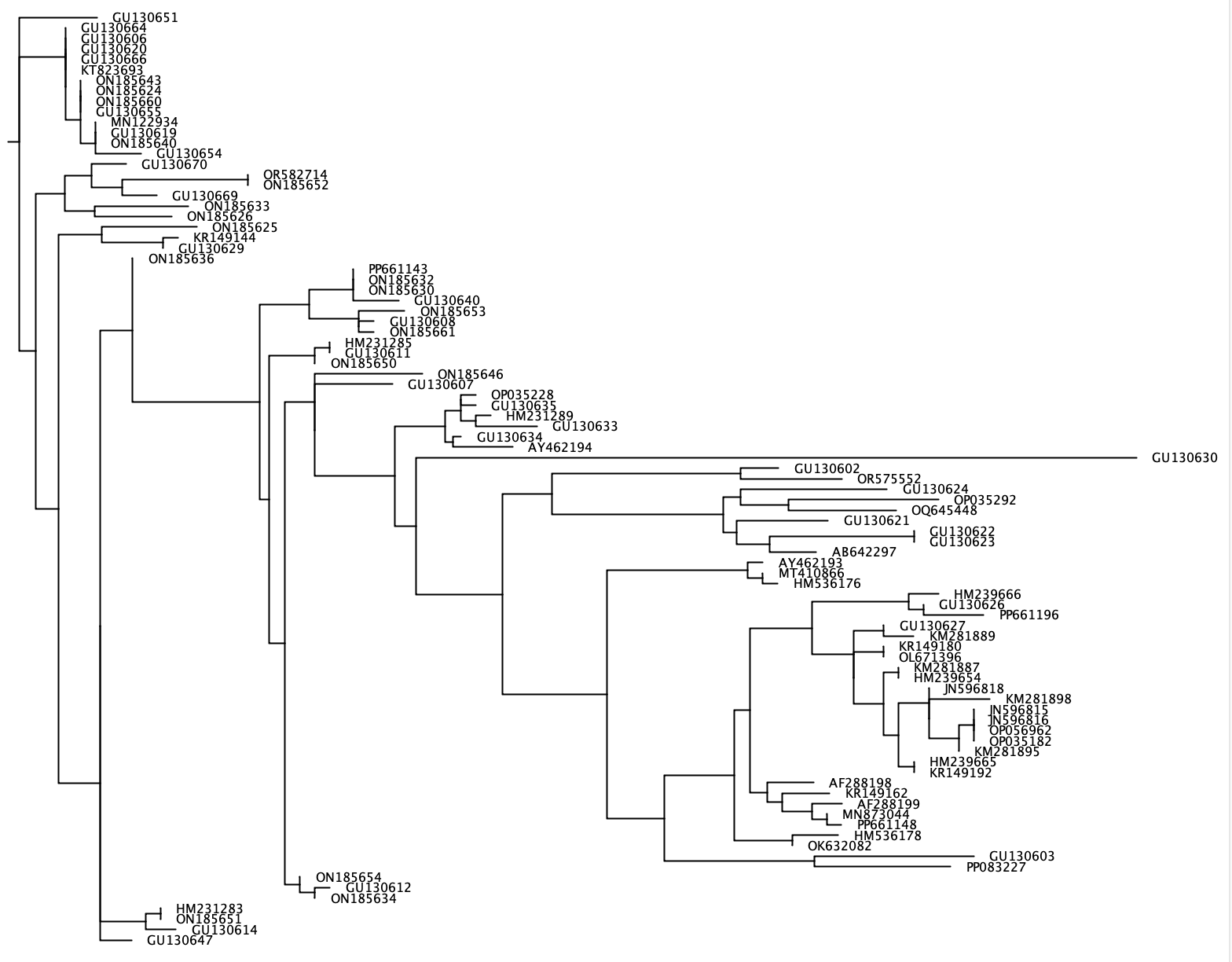
--phylogenetic-tree --phylogenetic-tree功能将生成一个系统发育树,以供有趣的物种列表。该感兴趣的物种可以使用--species参数导入,并由由+分隔的输入字符串或每个行上的单个物种名称组成。对于每种感兴趣的种类,将从参考数据库中提取序列,该数据库共享与感兴趣的物种共享用户定义的分类等级(参数: --tax-level )。螃蟹将使用clustalw2 v 2.1生成所有提取序列的比对,并使用FastTree生成一个邻里的系统发育树。这种以纽克格式的系统发育树将使用--output参数写入输出文件,并且可以在诸如Figtree或Geneious之类的软件程序中可视化。由于将为每种感兴趣的物种生成一个单独的系统发育树,因此--output输出以通用文件名为单位,而确切的输出文件将包含此通用名称,然后包含“ _species_name.tree”。
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
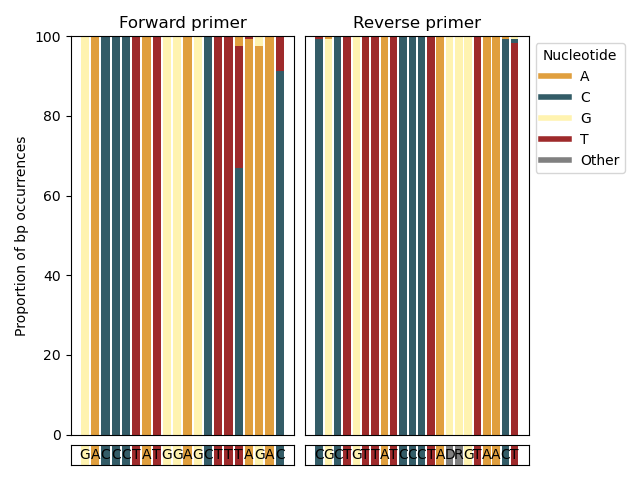
--amplification-efficiency-figure --amplification-efficiency-figure功能将产生条形图,以显示用户指定的分类学组的引物结合区域中的碱基对的比例,从而可视化向前和反向引物结合区域中的位置,其中不匹配的区域不匹配。可能发生在分类学群体中,可能会影响放大效率。使用--amplification-efficiency-figure参数将最终的Crabs形式--amplicons数据库作为输入,将最终的Crabs形式参考数据库作为输入。为了找到输入文件中每个序列的引物结合区域的信息,需要使用--input参数提供导入之后的最初下载的序列。使用--forward前向和--reverse反向参数提供了正向和反向引物序列(以5' - 3'方向)。可以使用--tax-group参数提供分类学群体的名称,并且可以在输入文件中包含的任何分类级别设置。最后,以.png格式的数字将写入--output参数指定的输出文件。
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table --completeness-table函数将输出一个选项卡式表(参数: --output ),并提供有关感兴趣物种列表的信息。该感兴趣的物种可以使用--species参数导入,并由由+分隔的输入字符串或每个行上的单个物种名称组成。使用--names和--nodes参数分别使用--download-taxonomy函数下载的“ names.dmp ”和“ nodes.dmp ”文件,将为每种感兴趣的种类生成分类谱系。输出表将有10列提供以下信息:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 :错误修复 - >在--import期间改进了大胆标头的解析。crabs --version v 1.0.5 :错误修复 - >根据BLAST+软件的需要,在构建BLAST数据库时向SEQ ID添加了一个长度限制。crabs --version v 1.0.4 :添加信息 - >提供了有关--pairwise-global-alignment --coverage --percent-identity的正确信息。crabs --version v 1.0.3 :错误修复 - >在中止分析前检查了3次NCBI服务器响应。crabs --version v 1.0.2 :错误修复 - >分析后返回0个序列时能够报告。crabs --version v 1.0.1 :错误修复 - >使用--species参数成功构建NCBI查询。

