reference_database_creator
bug fix --in-silico-pcr --untrimmed
螃蟹( C評論右參考資料庫一個複製子-乙阿塞德S equencing)是一種多功能軟體程序,可產生用於宏基因組分析的精選參考資料庫。 CRABS 工作流程由七個模組組成:(i)從線上儲存庫下載資料; (ii) 將下載的資料匯入CRABS格式; (iii) 以電腦PCR 分析萃取擴增子區域; (iv) 透過與電腦提取的條碼進行比對來檢索沒有引子結合區域的擴增子; (v) 透過多個篩選參數對本地資料庫進行管理和子集化; (vi) 根據分類器要求以各種格式匯出本機資料庫; (vi) 後處理功能,即可視覺化,以探索並提供本地參考資料庫的概要概述。這七個模組分為十八個功能,如下所述。此外,也為這十八個函數中的每一個函數提供了範例程式碼。最後,本 README 文件末尾提供了為 MiFish-E 引子集建立本地鯊魚參考資料庫的教程,以提供範例腳本供參考。
我們很高興地宣布,CRABS 根據用戶回饋進行了重大更新和程式碼重新設計,我們希望這將改善建立您自己的本地參考資料庫的使用者體驗!
請在下面找到 CRABS v 1.0.0中新增的功能和改進清單:
現在可以透過克隆此 GitHub 儲存庫來手動下載 CRABS v 1.0.0 (有關詳細信息,請參閱 4.1 手動安裝)。我們將盡快更新Docker容器和conda包,以方便安裝最新版本。
在您的研究專案中使用 CRABS 時,請引用以下論文:
[Jeunen, G.-J., Dowle, E., Edgecombe, J., von Ammon, U., Gemmell, N. J., & Cross, H. (2022). crabs—A software program to generate curated reference databases for metabarcoding sequencing data. Molecular Ecology Resources, 00, 1– 14.](https://doi.org/10.1111/1755-0998.13741)
CRABS 是一個運行在典型 Unix/Linux 環境上的僅命令列工具包,並且專門用 python3 編寫。然而,CRABS 利用 python 中的subprocess模組以 bash 語法運行多個命令,以規避 python 特定的特性並提高執行速度。我們提供了三種安裝 CRABS 的方法。對於最新版本的 CRABS,我們建議透過複製此 GitHub 儲存庫並分別安裝 10 個依賴項進行手動安裝(4.1 手動安裝中提供了所有依賴項的安裝說明)。 CRABS 也可以透過 Docker 和 conda 安裝。這兩種方法都可以透過自動共同安裝所有依賴項來輕鬆安裝。我們的目標是讓 Docker 容器和 conda 套件保持最新,儘管更新到最新版本可能會出現一定的延遲,尤其是 conda 套件。以下是所有三種方法的詳細資訊。
對於手動安裝,首先克隆 CRABS 儲存庫。此步驟要求 GitHub 可用於命令列(GitHub 的安裝說明)。
git clone https://github.com/gjeunen/reference_database_creator.git
根據您的設置,CRABS 可能需要在您的系統上可執行。這可以使用下面的程式碼來實現。
chmod +x reference_database_creator/crabs
安裝 CRABS 後,我們需要確保所有相依性均已安裝且可全域存取。最新版本的 CRABS(版本v 1.0.0 )在 Python 3.11.7(或 3.11.7 的任何相容版本)上運行,並依賴可能不是 Python 標準的五個 Python 模組以及五個外部軟體程式。下面列出了所有依賴項,以及安裝說明的連結。為每個模組和軟體程式提供的版本號碼是 CRABS 開發的版本號。儘管也可以使用每個版本的兼容版本。
Python 模組:
外部軟體程式:
安裝 CRABS 和所有依賴項後,可以使用下列程式碼在整個作業系統中存取 CRABS。
export PATH="/path/to/crabs/folder:$PATH"
將/path/to/crabs/folder替換為作業系統上 GitHub 儲存庫資料夾的實際路徑,即在上面的git clone指令期間所建立的資料夾。將export程式碼新增至.bash_profile或.bashrc 檔案中將使 CRABS 可以隨時全域存取。
Docker 是一個開源項目,允許在與電腦隔離的「容器」內部署軟體應用程序,並透過稱為 Docker Engine 的虛擬主機作業系統運作。與虛擬機器相比,執行 docker 的主要優點是它們使用的資源少得多。這種隔離意味著您可以在大多數作業系統上執行 Docker 容器,包括 Mac、Windows 和 Linux。您可能需要設定免費帳戶才能使用 Docker Desktop。此連結很好地介紹了使用 Docker 的基礎知識。以下連結可協助您入門並了解 Docker 多元宇宙。
只需兩個步驟即可讓 Crabs 在您的電腦上運作。首先,在您的電腦上安裝 Docker Desktop,這對大多數使用者來說是免費的。以下是 Mac 版的說明;這是針對 Windows 電腦的說明,這是針對 Linux 電腦的說明(支援大多數主要 Linux 平台)。一旦安裝並運行了 Docker Desktop(桌面應用程式必須運行,您才能在命令列上使用任何 docker 命令),您只需「拉取」我們的 Crabs 映像,就可以開始了:
docker pull quay.io/swordfish/crabs:0.1.7
雖然 Docker 應用程式的安裝很容易,但一開始使用這些應用程式可能會有點棘手。為了幫助您入門,我們提供了一些使用 Docker 版本的螃蟹的範例命令。這些範例可以在此儲存庫的 docker_intro 資料夾中找到。透過這些範例,您應該能夠完成整個參考資料庫的設定並做好準備。我們將繼續擴展這些範例,並在許多不同的情況下進行測試。請在“問題”選項卡中提出問題並提供回饋。
要安裝 conda 軟體包,必須先安裝 conda。請參閱此連結以了解詳細資訊。如果已安裝 conda,則最好在安裝 CRABS 之前使用conda update conda更新 conda 工具。
安裝 conda 後,請依照下列步驟安裝 CRABS 和所有相依性。確保按照下面顯示的順序輸入命令。
conda create -n CRABS
conda activate CRABS
conda config --add channels bioconda
conda config --add channels conda-forge
conda install -c bioconda crabs
輸入安裝命令後,conda 將處理請求(這可能需要一分鐘左右),然後顯示將安裝的所有軟體包和程序,並要求確認。輸入y開始安裝。完成後,CRABS 應該準備就緒。
我們已在 Mac 和 Linux 系統上測試了此安裝。我們尚未在適用於 Linux 的 Windows 子系統 (WSL) 上進行測試。
使用下面的代碼檢查CRABS是否安裝成功並拉取幫助資訊。
crabs -h幫助資訊將十八個函數分成不同的群組,每個群組在頂部列出該函數,在下面列出必需和可選的參數。

CRABS包含七個模組,包含十八個功能:
模組 1:從線上儲存庫下載數據
--download-taxonomy : 下載 NCBI 分類資訊;--download-bold :從生命條碼資料庫(BOLD)下載序列資料;--download-embl :從歐洲核苷酸檔案庫(ENA;EMBL)下載序列資料;--download-mitofish : 從MitoFish資料庫下載序列資料;--download-ncbi :從國家生物技術資訊中心(NCBI)下載序列資料。模組2:將下載的資料匯入CRABS格式
--import :將下載的序列或自訂條碼匯入為 CRABS 格式;--merge :將不同的 CRABS 格式的檔案合併到一個檔案中。模組 3:以電腦PCR 分析提取擴增子區域
--in-silico-pcr :透過定位和刪除引子結合區域從下載的資料中提取擴增子。模組 4:檢索沒有引子結合區域的擴增子
--pairwise-global-alignment :透過將下載的序列與電腦提取的條碼對齊來檢索沒有引子結合區域的擴增子。模組 5:透過多個過濾參數對本地資料庫進行管理和子集化
--dereplicate :丟棄重複序列;--filter : 透過多個過濾參數丟棄序列;--subset :對本機資料庫進行子集化以保留或排除指定的分類組。模組六:導出本地資料庫
--export :根據要使用的分類分類器的要求將 CRABS 格式的資料庫匯出為各種格式。模組 7:後處理功能,探索並提供本地參考資料庫的摘要概述
--diversity-figure :建立水平條形圖,顯示參考資料庫中包含的每個指定等級的物種和序列組的數量;--amplicon-length-figure :建立一個折線圖,描繪一個以分類組分隔的擴增子長度分佈;--phylogenetic-tree :使用目標物種清單的參考資料庫中的條碼建立系統發育樹;--amplification-efficiency-figure :建立一個長條圖,顯示引子結合區域中的不符情況;--completeness-table :建立包含分類組條碼可用性的電子表格。CRABS 可以從四個線上儲存庫下載初始定序數據,包括 (i) BOLD、(ii) EMBL、(iii) MitoFish 和 NCBI。從版本v 1.0.0開始,從每個儲存庫下載資料都分為自己的功能。另外,CRABS在下載數據後不會自動格式化數據,以增加靈活性,並在數據下載失敗時進行調試。
除了下載序列資料之外,CRABS 還能夠下載 NCBI 分類訊息,CRABS 使用該資訊為每個序列建立分類譜系。
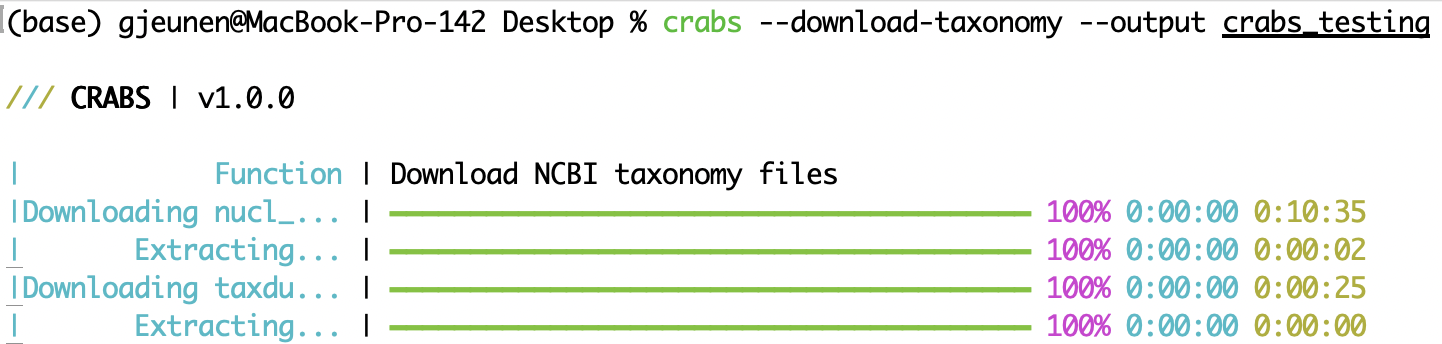
--download-taxonomy若要為參考資料庫中每個下載的序列指派分類譜系(請參閱 5.2 模組 2),需要下載分類資訊。 CRABS 利用 NCBI 的分類法並將三個特定文件下載到您的電腦:(i) 將登錄號連結到分類 ID 的文件 ( nucl_gb.accession2taxid ),(ii) 包含與每個分類ID 關聯的系統發育名稱的資訊的檔案 ( names.dmp ),以及 (iii) 包含分類 ID 如何連結資訊的檔案 ( nodes.dmp )。可以使用--output參數指定下載檔案的輸出目錄。若要排除檔案nucl_gb.accession2taxid或檔案names.dmp和nodes.dmp ,可以分別提供--exclude acc2tax或--exclude taxdump參數。下面的第一個程式碼不會下載任何文件,因為acc2tax和taxdump都是為--exclude參數提供的。第二行程式碼將所有三個檔案下載到子目錄--output crabs_testing 。下面的螢幕截圖顯示了執行這行程式碼時列印到控制台的內容。
crabs --download-taxonomy --exclude 'acc2taxid,taxdump'
crabs --download-taxonomy --output crabs_testing
--download-bold BOLD 序列透過 BOLD 網站下載。輸出文件的結構為兩行 fasta 文檔,可使用--output參數指定。使用者可以使用--taxon參數指定要下載的分類組。當使用者想要下載多個分類組時,我們建議編寫一個簡單的 for 迴圈(下面提供的範例),從而限制每個實例從 BOLD 下載的資料量。但是,如果僅對有限數量的分類組感興趣,則分類組名稱也可以用|分隔。 (下面提供的範例)。我們也建議使用者檢查要下載的分類群組名稱是否在粗體存檔中列出,或是否需要使用替代名稱。例如,指定--taxon Chondrichthyes將不會從 BOLD 下載所有軟骨魚類序列,因為該類別名稱未在 BOLD 中列出。在這種情況下,使用者應該使用--taxon Elasmobranchii 。使用者也可以透過提供--marker參數來指定將下載限制為特定的遺傳標記。當對多個遺傳標記感興趣時,標記名稱應以|分隔。 。 BOLD 上的四個主要 DNA 條碼標記是COI-5P 、 ITS 、 matK和rbcL 。 --marker參數的輸入區分大小寫。
建議方法:一個簡單的 for 循環,用於從 BOLD 下載多個分類組的資料(建議方法)。下面的程式碼首先下載板鰓亞綱的數據,然後下載分配給哺乳動物的序列。下載的資料將寫入子目錄--output crabs_testing並放置在兩個單獨的檔案中,指示哪些資料屬於哪個分類組,即crabs_testing/bold_Elasmobranchii.fasta和crabs_testing/bold_Mammalia.fasta 。
for taxon in Elasmobranchii Mammalia; do crabs --download-bold --taxon ${taxon} --output crabs_testing/bold_${taxon}.fasta; done

替代選項:除了建議的 for 迴圈之外,還可以透過使用|分隔名稱來一次提供多個分類單元名稱。 。
crabs --download-bold --taxon 'Elasmobranchii|Mammalia' --output crabs_testing/bold_elasmobranchii_mammalia.fasta
--download-embl EMBL 的序列可透過 ENA FTP 網站下載。 EMBL 檔案會先以「.fasta.gz」格式下載,下載完成後會自動解壓縮。與 BOLD 或 NCBI 相比,該資料庫在選擇性下載方面沒有提供足夠的靈活性。相反,EMBL 資料分為 15 個稅務部門,可以單獨下載。可以使用--taxon參數指定要下載的稅務部門。由於每個稅務部門都分為多個文件,因此在名稱後面提供*以下載所有文件。用戶還可以透過寫下完整的文件名來下載特定文件。下面提供了所有 15 個稅務劃分選項的清單。可以使用--output參數指定輸出目錄和檔名。
稅務部門列表:
crabs --download-embl --taxon 'mam*' --output crabs_testing/embl_mam.fasta
--download-mitofish CRABS還可以下載MitoFish資料庫。這個資料庫是一個兩行的 fasta 檔案。可以使用--output參數指定輸出目錄和檔名。
crabs --download-mitofish --output crabs_testing/mitofish.fasta

--download-ncbi NCBI 資料庫中的序列透過 Entrez 程式設計實用程式下載。 NCBI 允許從各種資料庫下載數據,使用者可以使用--database參數指定。對於大多數使用者來說, --database nucleotide資料庫最適合建立本地參考資料庫。
若要指定要從 NCBI 下載的數據,使用者可以透過--query參數提供搜尋。精心設計 NCBI 檢索可能很困難。建立搜尋查詢的一個好方法是使用 NCBI 網頁搜尋視窗。從此連結中,首先進行初始搜尋並按 Enter 鍵。這將帶您進入結果頁面,您可以在其中進一步優化搜尋。在下面的螢幕截圖中,我們透過將序列長度限制在 100 - 25,000 bp 之間並且僅包含粒線體序列來進一步細化搜尋。使用者可以將文字複製並貼上到網站上的「搜尋詳細資料」方塊中,並將其放在引號中提供給--query參數。使用 NCBI 網頁搜尋視窗的另一個好處是,網頁將顯示有多少序列與您的搜尋查詢匹配,這應該與 CRABS 報告的序列數量相符。此網頁提供了有關使用 NCBI 網頁上的搜尋功能的進一步簡短教程,該教程是我們團隊為額外資訊而編寫的。
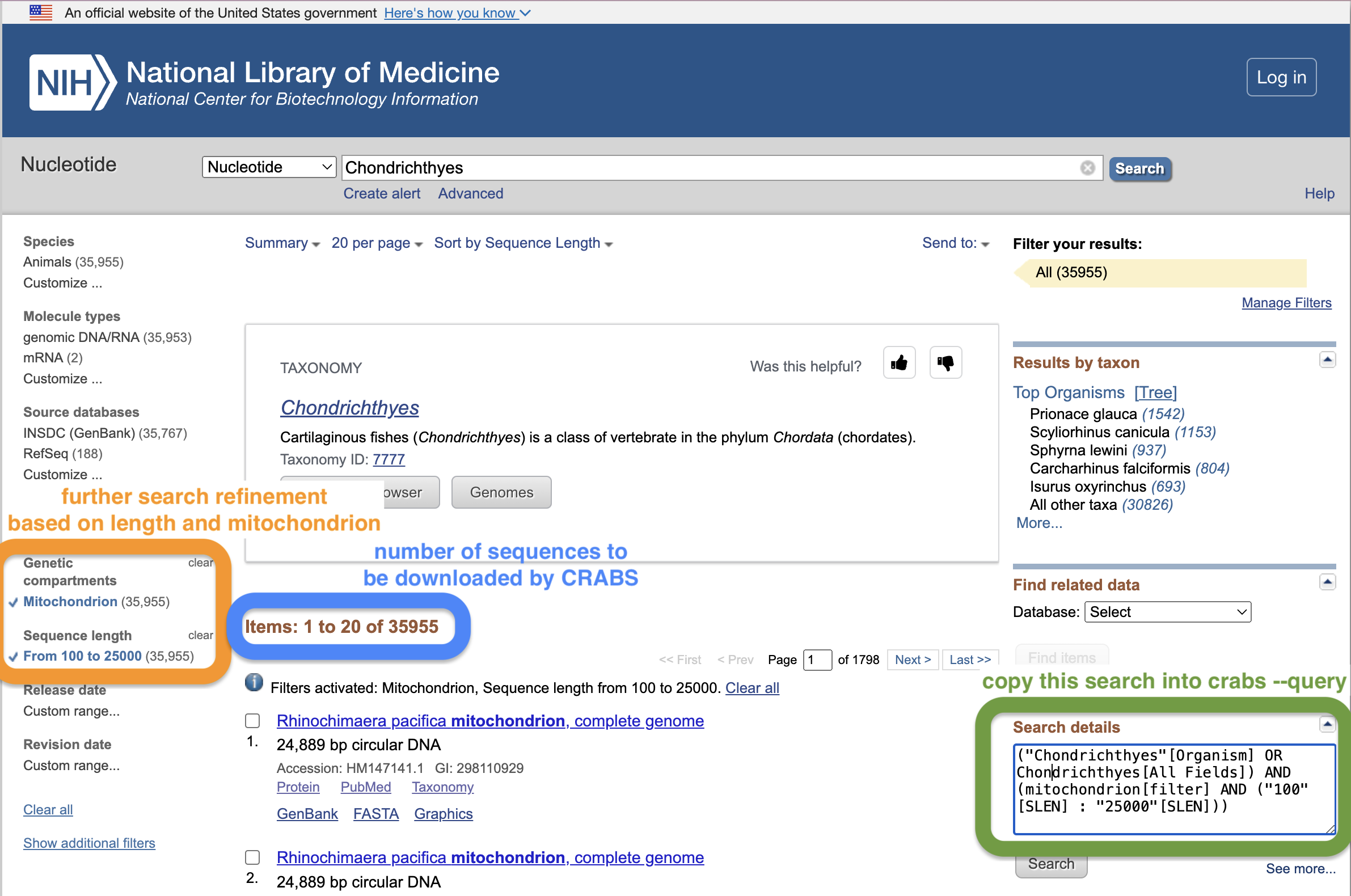
除了搜尋查詢 ( --query ) 之外,使用者還可以使用--species參數下載物種清單的序列數據,進一步限制搜尋項。 --species參數要麼採用由+分隔的物種名稱輸入字串,要麼採用文件中每行一個物種名稱的輸入 .txt 檔案。 --batchsize參數為使用者提供從 NCBI 網站批次下載 N 序列的選項。此參數預設為 5,000。不建議將此值增加到 5,000 以上,因為如果一次下載太多序列,NCBI 伺服器很可能會中斷下載。 --email參數允許使用者指定其電子郵件地址,這是訪問 NCBI 伺服器所必需的。最後,可以使用--output參數指定輸出目錄和檔名。
crabs --download-ncbi --query '("Chondrichthyes"[Organism] OR Chondrichthyes[All Fields]) AND (mitochondrion[filter] AND ("100"[SLEN] : "25000"[SLEN]))' --output crabs_testing/ncbi_chondrichthyes.fasta --email [email protected] --database nucleotide

--import下載線上儲存庫中的資料後,需要使用--import函數將檔案匯入 CRABS 中。 CRABS 格式構成每個序列的單一製表符分隔行,包含所有信息,包括(i) 序列ID、(ii) 從初始下載解析的分類名稱、(iii) NCBI 分類單元ID 號、(iv) 根據NCBI分類的分類譜系,和(v)序列。 CRABS 將嘗試取得每個序列的 NCBI 登錄號作為序列 ID。如果序列不包含登錄號,即未保藏在 NCBI 上,CRABS 將使用下列格式產生唯一的序列 ID: crabs_*[num]*_taxonomic_name 。輸入文件的格式使用--import-format參數指定,並指定從中下載資料的儲存庫的名稱,即BOLD 、 EMBL 、 MITOFISH或NCBI 。 CRABS 創建的分類譜系基於 NCBI 分類,並且 CRABS 需要使用--download-taxonomy函數下載的三個文件,即--names 、 --nodes和--acc2tax 。從版本v 1.0.0開始,CRABS 能夠解析同義詞和不可接受的名稱,以將大量序列和多樣性納入本地參考資料庫中。可以使用--ranks參數指定要包含在分類譜系中的分類等級。雖然可以包含任何分類排名,但我們建議使用以下輸入來包含大多數分類分類器的所有必要資訊--ranks 'superkingdom;phylum;class;order;family;genus;species' 。輸出檔案可以使用--output參數指定,並且是一個簡單的 .txt 檔案。在終端視窗中,CRABS 列印導入序列數量的結果,以及無法產生分類譜系的任何序列。
crabs --import --import-format bold --input crabs_testing/bold_Elasmobranchii.fasta --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --acc2tax crabs_testing/nucl_gb.accession2taxid --output crabs_testing/crabs_bold.txt --ranks 'superkingdom;phylum;class;order;family;genus;species'

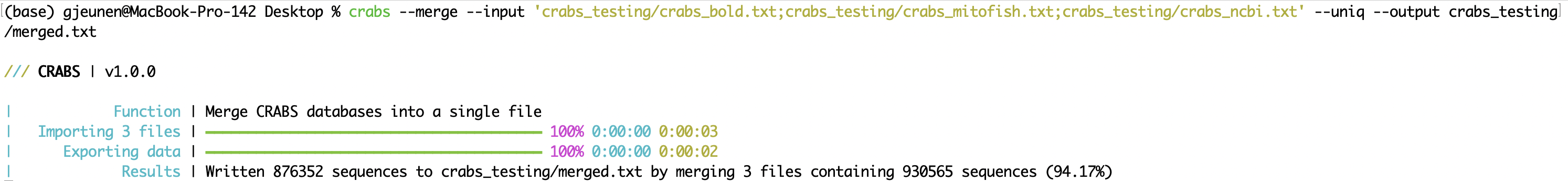
--merge當從多個線上儲存庫下載序列資料時,可以使用--merge函數在匯入後將檔案合併為單一檔案(請參閱 5.2.1 --import )。可以使用--input參數輸入要合併的輸入文件,文件之間以; 。一個序列在存放到各個線上儲存庫時可能會被多次下載。使用--uniq參數僅保留每個入藏號的單一版本。可以使用--output參數指定輸出檔。在終端機視窗中,CRABS 列印合併序列數的結果,以及使用--uniq參數時保留的序列數。
crabs --merge --input 'crabs_testing/crabs_bold.txt;crabs_testing/crabs_mitofish.txt;crabs_testing/crabs_ncbi.txt' --uniq --output crabs_testing/merged.txt
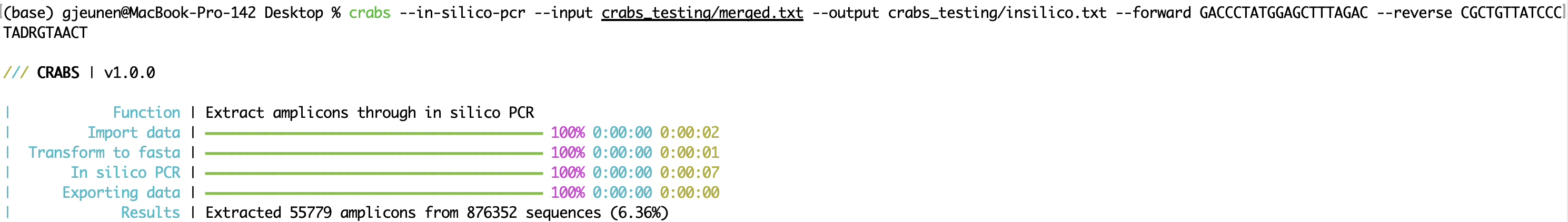
CRABS 透過進行電腦PCR(功能: --in-silico-pcr )萃取引子組的擴增子區域。 CRABS 使用 cutadapt v 4.4進行電腦PCR,以提高傳統 Python 程式碼的執行速度。可以分別使用“ --input ”和“ --output ”參數指定輸入和輸出檔名。正向和反向引子應分別使用「 --forward 」和「 --reverse 」參數在 5'-3' 方向提供。 CRABS 將反向互補反向引子。從版本v 1.0.0開始,CRABS 能夠使用單一電腦PCR 分析在兩個方向上保留條碼。因此,無需進行反向互補步驟和重新運行電腦PCR,從而顯著提高執行速度。若要保留找不到引子結合區域的序列,可以為--untrimmed參數指定輸出檔。可以使用--mismatch參數指定引子結合區域中發現的最大允許錯配數,預設為4 。預設使用最大執行緒數,但使用者可以使用--threads參數指定要使用的執行緒數。
crabs --in-silico-pcr --input crabs_testing/merged.txt --output crabs_testing/insilico.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT
當將參考序列存入線上資料庫時,通常的做法是從參考序列中刪除引子結合區域。因此,當使用與--in-silico-pcr函數中搜尋的相同正向和/或反向引子產生參考序列時, --in-silico-pcr函數將無法恢復該序列的擴增子區域。序列。為了解決這種可能性,CRABS 可以選擇運行使用 VSEARCH v 2.16.0實現的成對全局比對,以提取參考序列不包含完整正向和反向引子結合區域的擴增子區域。為了實現這一點, --pairwise-global-alignment函數使用--input參數接收最初下載的資料庫檔案。要搜尋的資料庫是--in-silico-pcr的輸出文件,可以使用--amplicons參數指定。可以使用--output參數指定輸出檔。引子序列僅用於計算鹼基對長度,可使用--forward和--reverse參數進行設定。由於--pairwise-global-alignment函數對於大型資料庫可能需要很長時間才能運行,因此可以使用--size-select參數限制序列長度以加快該過程。可以分別使用--percent-identity和--coverage參數指定最小百分比身分和查詢覆蓋率。 --percent-identity應以 0 到 1 之間的百分比值形式提供(例如,95% = 0.95),而--coverage應以0 到100 之間的百分比值形式提供(例如,95% = 95) 。預設情況下, --pairwise-global-alignment函數僅限於保留引子序列未完全存在於參考序列中的序列(比對在正向或反向引子的長度內開始或結束)。當提供--all-start-positions參數時,當發現比對超出引子結合區域範圍時,將包含正命中(由於--in-silico-pcr函數由於引子結合區域中有太多不匹配而錯過)引子結合區)。我們不建議使用--all-start-positions ,因為當引子中存在超過 4 個不匹配時,使用--in-silico-pcr函數的指定引子組擴增條碼的可能性很小 -結合區域。
crabs --pairwise-global-alignment --input crabs_testing/merged.txt --amplicons crabs_testing/insilico.txt --output crabs_testing/aligned.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --size-select 10000 --percent-identity 0.95 --coverage 0.95
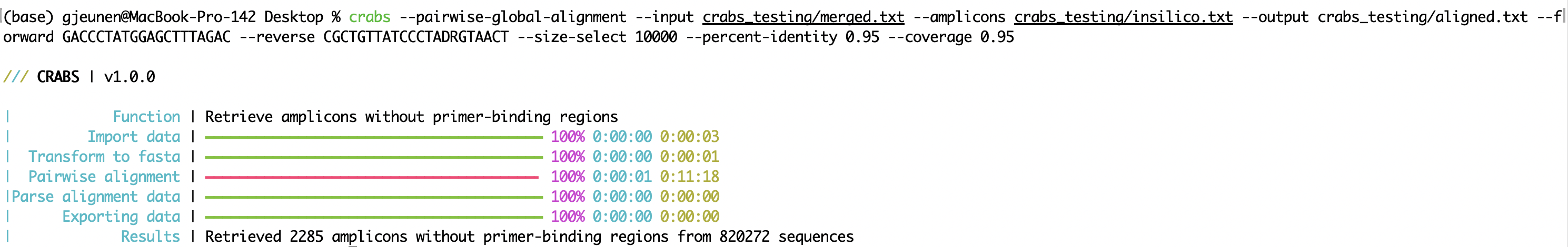
--pairwise-global-alignment的程式碼執行速度當 CRABS 處理大型序列檔案時,即使支援多線程, --pairwise-global-alignment函數也可能需要花費大量時間來執行。自從更新到 CRABS v 1.0.0以來,從--import到--export都有相同的檔案結構,從而使函數能夠以任何順序執行。雖然我們仍然建議遵循 CRABS 工作流程的順序,但在--in-silico-pcr函數之前執行--dereplicate和--filter函數時,可以顯著加快--pairwise-global-alignment函數的速度。透過在--in-silico-pcr之前執行這些管理步驟,CRABS 為--pairwise-global-alignment函數處理所需的序列數量將顯著減少。
注意 1 :在--in-silico-pcr之前執行--filter函數時,請確保省略任何直接影響序列的參數,因為--filter會基於整個序列而不是提取的擴增子。因此,省略以下參數: --minimum-length 、 --maximum-length 、 --maximum-n 。
註 2 :在--in-silico-pcr之前執行--dereplicate和--filter函數時,建議在--pairwise-global-alignment之後再次運行這兩個函數,因為現在可以進一步整理資料庫擴增子被提取。
一旦透過--in-silico-pcr和--pairwise-global-alignment函數提取了引子集的所有潛在條碼,本地參考資料庫就可以使用各種函數(包括--dereplicate在CRABS 中進行進一步的整理和子集化、 --filter和--subset 。
--dereplicate第一種管理方法是使用--dereplicate函數取消複製本機參考資料庫。對於某些分類單元,此時本機參考資料庫中可能包含多個相同的條碼。當不同的研究小組沉積了相同的序列,或者如果提取的條碼中不包含分類單元序列之間的特異性內變異,則可能會發生這種情況。最好刪除這些相同的參考條碼以加速分類分配,並改善分類分配結果(特別是對於提供有限數量結果的分類分類器,即BLAST)。
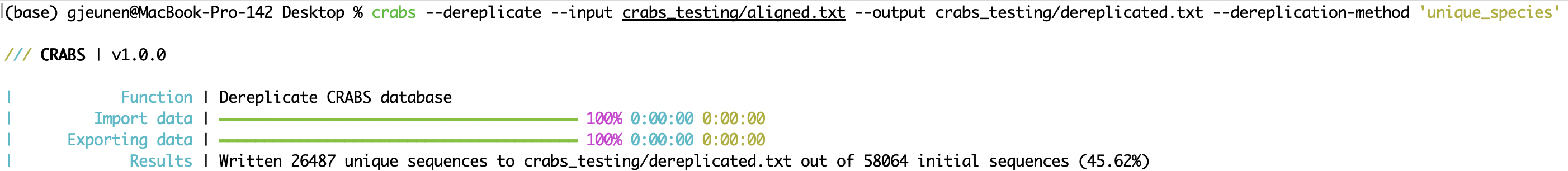
可以分別使用--input和--output參數指定輸入和輸出檔。 CRABS提供了三種去複製方法,可以使用--dereplication-method參數指定,包括:
crabs --dereplicate --input crabs_testing/aligned.txt --output crabs_testing/dereplicated.txt --dereplication-method 'unique_species'
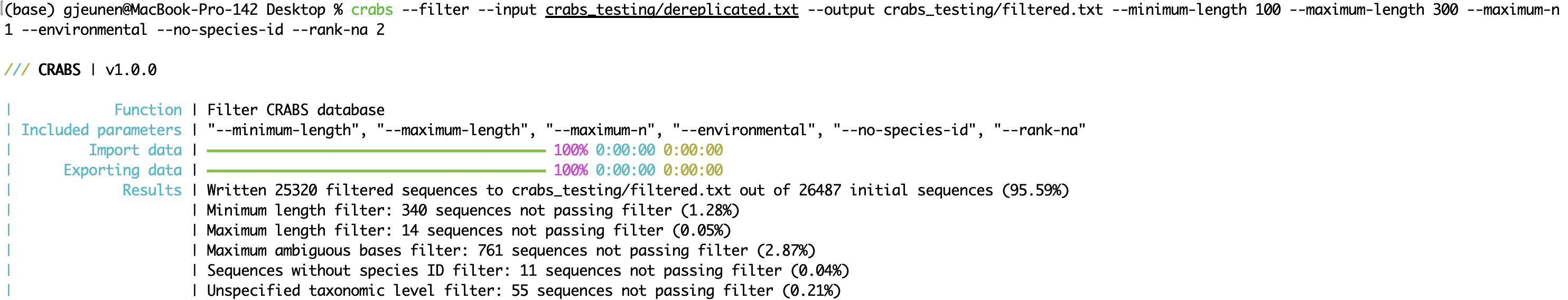
--filter第二種管理方法是使用--filter函數使用各種參數來過濾本機參考資料庫。可以分別使用--input和--output參數指定輸入和輸出檔。從版本v 1.0.0開始。 CRABS 結合了基於六個參數的過濾,包括:
--minimum-length :資料庫中保留的擴增子的最小序列長度;--maximum-length :資料庫中保留的擴增子的最大序列長度;--maximum-n : 丟棄具有 N 個或更多不明確鹼基 ( N ) 的擴增子;--environmental :從資料庫中丟棄環境序列;--no-species-id :丟棄沒有物種名稱的序列;--rank-na :丟棄具有 N 個或更多未指定分類等級的序列。 crabs --filter --input crabs_testing/dereplicated.txt --output crabs_testing/filtered.txt --minimum-length 100 --maximum-length 300 --maximum-n 1 --environmental --no-species-id --rank-na 2
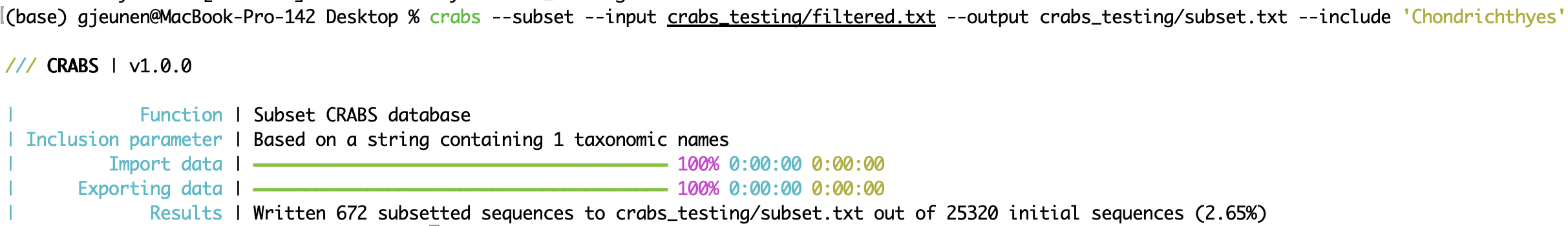
--subset CRABS 中包含的第三種也是最後一種管理方法是使用--subset函數對本地參考資料庫進行子集化,以包含(參數: --include )或排除(參數: --exclude )特定分類單元。此功能允許從與研究問題不相關的分類組中刪除參考條碼。由於引子組潛在的非特異性擴增,這些分類組可能已納入當地參考資料庫。 --subset的另一個用例是刪除已知的錯誤序列。
對於基於機器學習(IDTAXA) 或k-mer 距離(SINTAX) 的分類學分類器,透過僅包含已知在採樣區域中出現的類群並排除未知的密切相關物種來對參考資料庫進行子集化可能是有益的發生在該區域以增加這些分類器獲得的分類分辨率並獲得改進的分類分配結果。
可以分別使用--input和--output參數指定輸入和輸出檔。 --include和--exclude參數可以接受由 ; 分隔的分類群列表;或每行包含一個分類單元名稱的 .txt 檔案。
crabs --subset --input crabs_testing/filtered.txt --output crabs_testing/subset.txt --include 'Chondrichthyes'
參考資料庫最終確定後,可以將其匯出為各種格式,以適應大多數將分類法分配給宏基因組資料的軟體工具所需的規格。可以分別使用--input和--output參數指定輸入和輸出檔。從版本v 1.0.0開始,CRABS 合併了六種不同分類器的參考資料庫格式(參數: --export-format ),包括:
--export-format 'sintax' : SINTAX 分類器合併到 USEARCH 和 VSEARCH 中;--export-format 'rdp' :RDP 分類器是一個廣泛用於微生物組研究的獨立程式;--export-format 'qiime-fasta'和--export-format 'qiime-text' :可用於在 QIIME 和 QIIME2 中分配分類 ID;--export-format 'dada2-species'和--export-format 'dada2-taxonomy' :可用於在 DADA2 中分配分類 ID;--export-format 'idt-fasta'和--export-format 'idt-text' :IDTAXA 分類器是包含在 DECIPHER R 套件中的機器學習演算法;--export-format 'blast-notax' :為blastn和megablast建立本機BLAST參考資料庫,其中輸出不提供分類ID,但列出入藏號;--export-format 'blast-tax' :為blastn和megablast建立本機BLAST參考資料庫,其中輸出提供分類ID和登錄號。 crabs --export --input crabs_testing/subset.txt --output crabs_testing/BLAST_TAX_CHONDRICHTHYES --export-format 'blast-tax'

雖然將本地參考資料庫匯出為單一格式(參考資料庫分為多個文件的分類器除外,即QIIME、DADA2、IDTAXA)對於大多數用戶來說就足夠了,但可以編寫一個簡單的for 循環來導出本地參考資料庫如果使用者想要比較不同分類器之間的結果,請使用多種格式的參考資料庫。下面提供了以 SINTAX、RDP 和 IDTAXA 格式匯出本機參考資料庫的範例。
for format in sintax.fasta rdp.fasta idt-fasta.fasta idt-text.txt; do crabs --export --input crabs_testing/subset.txt --output crabs_testing/chondrichthyes_${format} --export-format ${format%%.*}; done

一旦參考資料庫最終確定,CRABS 可以運行五個後處理函數來探索並提供本地參考資料庫的摘要概述,包括 (i) --diversity-figure 、(ii) --amplicon-length-figure 、( iii) --phylogenetic-tree ,(iv) --amplification-efficiency-figure ,和(v) --completeness-table 。
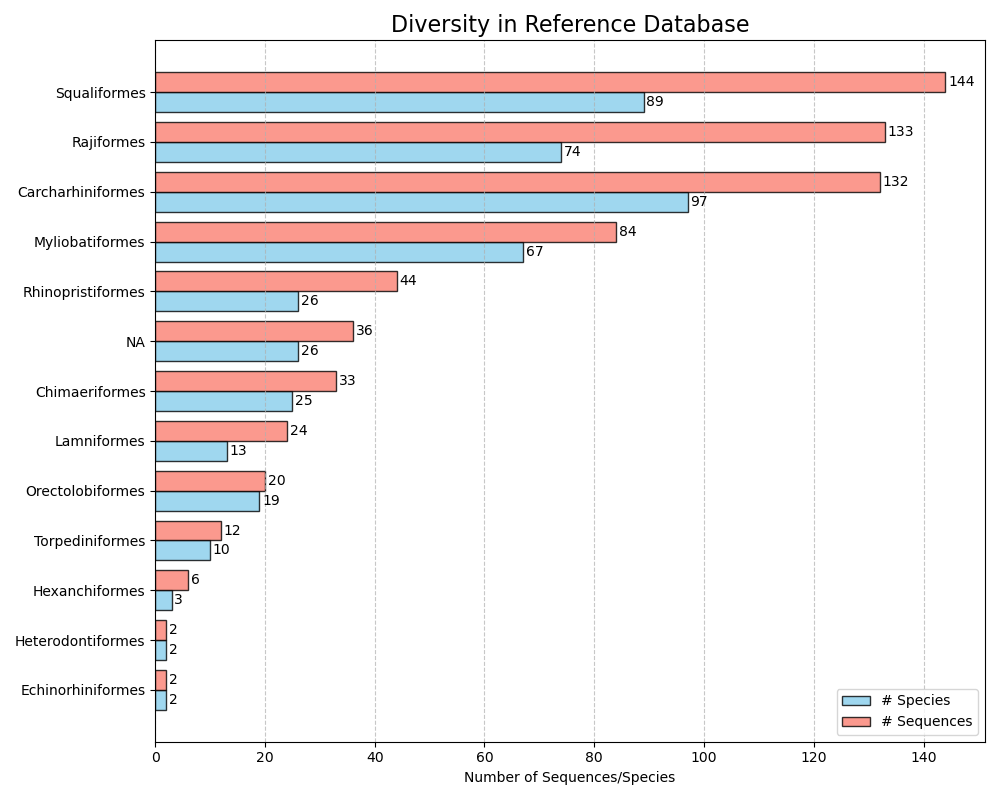
--diversity-figure --diversity-figure函數產生一個水平條形圖,其中包含參考資料庫中每個分類組的物種數(藍色)和序列數(橘色)。使用者可以使用--tax-level參數指定分類等級來分割參考資料庫。稅收等級是在--import函數中出現的等級編號。例如,如果在基於 superkingdom 的--import拆分期間使用--ranks 'superkingdom;phylum;class;order;family;genus;species'則需要--tax-level 1 , phylum = --tax-level 2 , class = --input --tax-level 3等。 .png 格式的圖形將寫入輸出文件,可以使用--output參數指定該文件。
crabs --diversity-figure --input crabs_testing/subset.txt --output crabs_testing/diversity-figure.png --tax-level 4
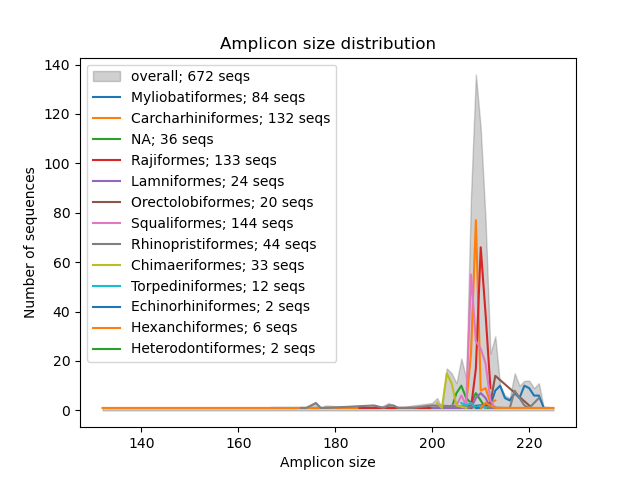
--amplicon-length-figure --amplicon-length-figure函數產生顯示擴增子長度範圍的折線圖。參考資料庫中所有序列的擴增子長度的總體範圍以灰色陰影顯示,而每個分類組(參數: --tax-level )分割的結果由彩色線覆蓋。此外,圖例還顯示分配給每個分類組的序列數以及參考資料庫中的序列總數。可以使用--input參數指定CRABS格式的輸入檔。 .png 格式的圖形將寫入輸出文件,可以使用--output參數指定該文件。
crabs --amplicon-length-figure --input crabs_testing/subset.txt --output crabs_testing/amplicon-length-figure.png --tax-level 4
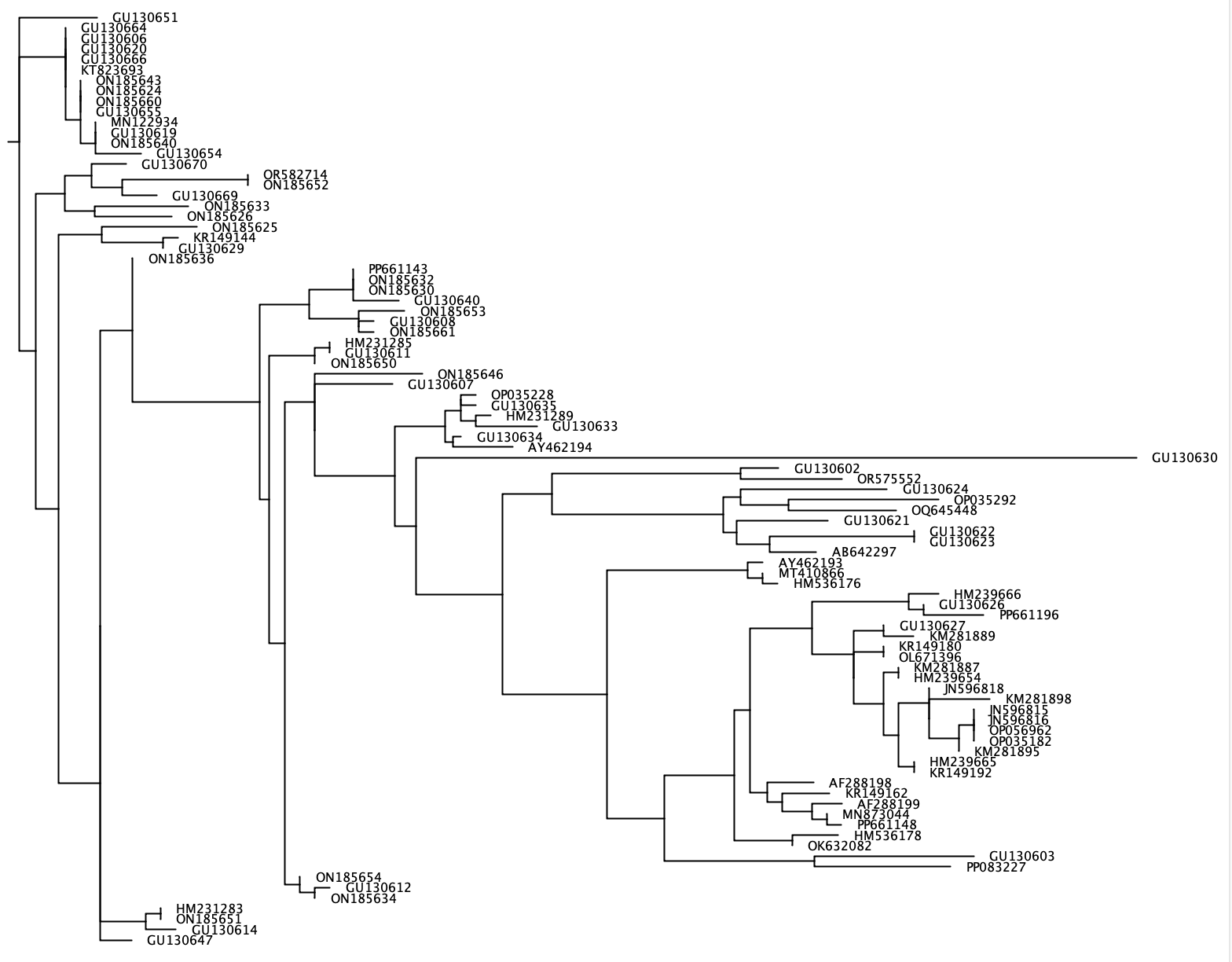
--phylogenetic-tree --phylogenetic-tree函數將為感興趣的物種清單產生系統發育樹。可以使用--species參數導入此感興趣物種列表,並由用+分隔的輸入字串或每行包含單一物種名稱的 .txt 檔案組成。對於每個感興趣的物種,將從參考資料庫中提取序列,該序列與感興趣的物種共享用戶定義的分類等級(參數: --tax-level )。 CRABS 將使用 clustalw2 v 2.1 產生所有萃取序列的比對,並使用 FastTree 產生鄰接系統發育樹。 newick 格式的系統發育樹將使用--output參數寫入輸出文件,並且可以在 FigTree 或 Geneious 等軟體程式中進行視覺化。由於將為每個感興趣的物種生成單獨的系統發育樹,因此--output採用通用文件名,而確切的輸出文件將包含此通用名稱,後跟“_species_name.tree”。
crabs --phylogenetic-tree --input crabs_testing/subset.txt --output crabs_testing/phylo --tax-level 4 --species 'Carcharodon carcharias+Squalus acanthias'
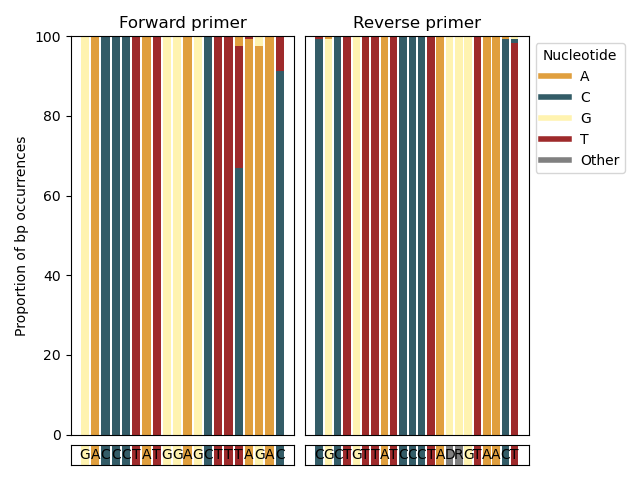
--amplification-efficiency-figure --amplification-efficiency-figure函數將產生一個條形圖,顯示使用者指定的分類組的引子結合區域中鹼基對出現的比例,從而可視化正向和反向引子結合區域中不匹配的位置可能發生在感興趣的分類群中,可能影響擴增效率。 --amplification-efficiency-figure函數使用--amplicons參數將最終 CRABS 格式的參考資料庫作為輸入。要查找輸入檔中每個序列的引子結合區域的信息,需要使用--input參數提供導入後最初下載的序列。使用--forward和--reverse參數提供正向和反向引子序列(5' - 3' 方向)。可以使用--tax-group參數提供感興趣的分類群組的名稱,並且可以在輸入檔案中合併的任何分類層級進行設定。最後,.png格式的圖形將會寫入--output參數指定的輸出檔。
crabs --amplification-efficiency-figure --input crabs_testing/merged.txt --amplicons crabs_testing/subset.txt --forward GACCCTATGGAGCTTTAGAC --reverse CGCTGTTATCCCTADRGTAACT --output crabs_testing/amplification-efficiency.png --tax-group Carcharhiniformes
--completeness-table --completeness-table函數將輸出一個製表符分隔的表(參數: --output ),其中包含有關感興趣物種列表的資訊。可以使用--species參數導入感興趣的物種列表,該列表包含以+分隔的輸入字串或每行包含單一物種名稱的 .txt 檔案。將使用分別使用--names和--nodes參數使用--download-taxonomy函數下載的「 names.dmp 」和「 nodes.dmp 」檔案為每個感興趣的物種產生分類譜系。輸出表將有 10 列,提供以下資訊:
crabs --completeness-table --input crabs_testing/subset.txt --output crabs_testing/completeness.txt --names crabs_testing/names.dmp --nodes crabs_testing/nodes.dmp --species 'Carcharodon carcharias+Squalus acanthias'
crabs --version v 1.0.6 :錯誤修復 --> 改進了--import期間對 BOLD 標頭的解析。crabs --version v 1.0.5 :錯誤修復 --> 根據 BLAST+ 軟體的需要,在建立 BLAST 資料庫時添加了 seq ID 的長度限制。crabs --version v 1.0.4 :新增了資訊 --> 為--pairwise-global-alignment --coverage --percent-identity的值輸入提供了正確的資訊。crabs --version v 1.0.3 :錯誤修復 --> 在中止分析之前檢查 NCBI 伺服器回應 3 次。crabs --version v 1.0.2 :錯誤修復 --> 能夠在分析後傳回 0 個序列時進行報告。crabs --version v 1.0.1 :錯誤修復 --> 使用--species參數成功建立 NCBI 查詢。

