يحتوي هذا الريبو على:
يتطلب sepal python3 ، ويفضل أن يكون إصدارًا أحدث من أو يساوي 3.5. للتنزيل والتثبيت، افتح الوحدة الطرفية وقم بالتغيير إلى الدليل الذي تريد تنزيل sepal إليه وقم بما يلي:
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
اعتمادًا على امتيازات المستخدم لديك، قد يتعين عليك إضافة --user كوسيطة إلى setup.py . سيمنحك تشغيل الإعداد الحد الأدنى المطلوب من التثبيت لحساب أوقات الانتشار. ومع ذلك، إذا كنت تريد أن تكون قادرًا على استخدام وحدات التحليل، فستحتاج أيضًا إلى تثبيت الحزم الموصى بها. للقيام بذلك، ببساطة (في نفس الدليل) قم بتشغيل:
pip install -e " .[full] " مرة أخرى، قد يكون من الضروري تضمين --user . أيضًا، قد تضطر إلى استخدام pip3 إذا كانت هذه هي الطريقة التي قمت بها بإعداد واجهة python-pip . إذا كنت تستخدم البيئات الافتراضية أو conda ، فاتبع توصياتهم لتثبيت الحزم.
يجب أن يؤدي هذا إلى تثبيت واجهة سطر الأوامر (CLI) والحزمة القياسية. لاختبار ومعرفة ما إذا كان التثبيت ناجحًا، يمكنك محاولة تنفيذ الأمر:
sepal -h
والتي يجب أن تطبع رسالة المساعدة المرتبطة بـ sepal. إذا نجح كل شيء بالنسبة لك حتى الآن، فيمكنك المتابعة إلى قسم الأمثلة لرؤية sepal أثناء العمل!
الاستخدام الموصى به لـ sepal هو من خلال واجهة سطر الأوامر. يمكن بسهولة إجراء كل من عمليات المحاكاة من أجل حساب أوقات الانتشار بالإضافة إلى التحليل اللاحق أو فحص النتائج عن طريق كتابة sepal متبوعًا إما run أو analyze . تحتوي وحدة analyze على خيارات مختلفة، لتصور النتائج ( inspect )، أو فرز الملفات الشخصية إلى عائلات نمطية ( family ) أو إخضاع العائلات المحددة لتحليل الإثراء الوظيفي ( fea ). للحصول على قائمة كاملة بالأوامر المتاحة، قم بتنفيذ sepal module -h ، حيث الوحدة هي إحدى وحدات run analyze . نوضح أدناه كيف يمكن استخدام sepal للعثور على ملفات تعريف النسخ ذات الأنماط المكانية.
سنقوم بإنشاء مجلد للاحتفاظ بنتائجنا، والذي سيظهر أيضًا كدليل العمل الخاص بنا. من الدليل الرئيسي للريبو، قم بما يلي:
cd res
mkdir example
cd exampleسيتم استخدام عينة MOB لتجسيد تحليلنا. نبدأ بحساب أوقات الانتشار لكل ملف تعريف النسخ:
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1فيما يلي مثال (مع عرض إضافي لأمر المساعدة) لكيفية ظهور ذلك

بعد حساب أوقات الانتشار، نريد فحص النتيجة، كما هو الحال في الدراسة، سننظر إلى أفضل 20 ملفًا شخصيًا. يمكننا بسهولة إنشاء صور من نتائجنا عن طريق تشغيل الأمر:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5والذي سيبدو شيئًا في سطر هذا:

وسيكون الإخراج الصورة التالية:
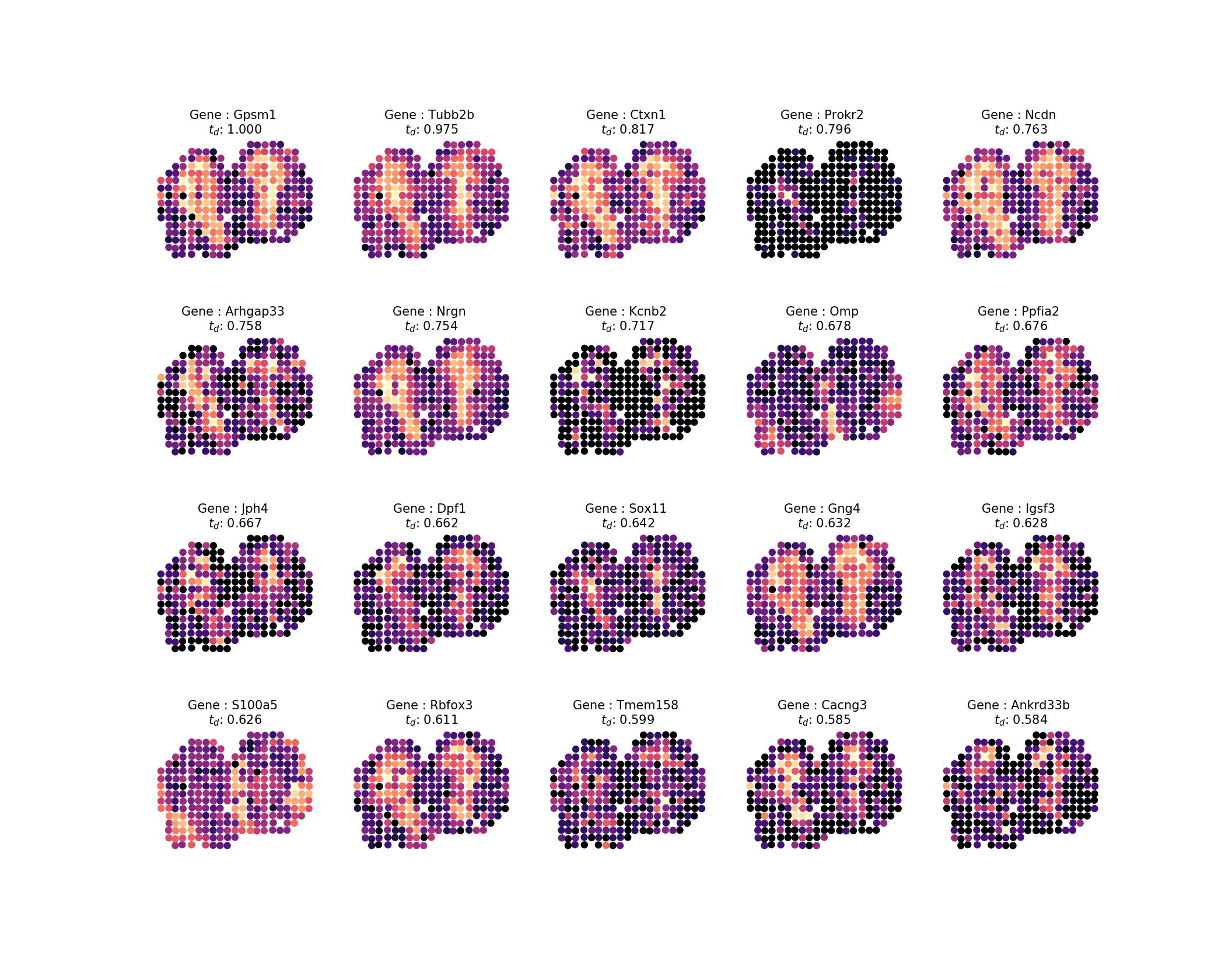
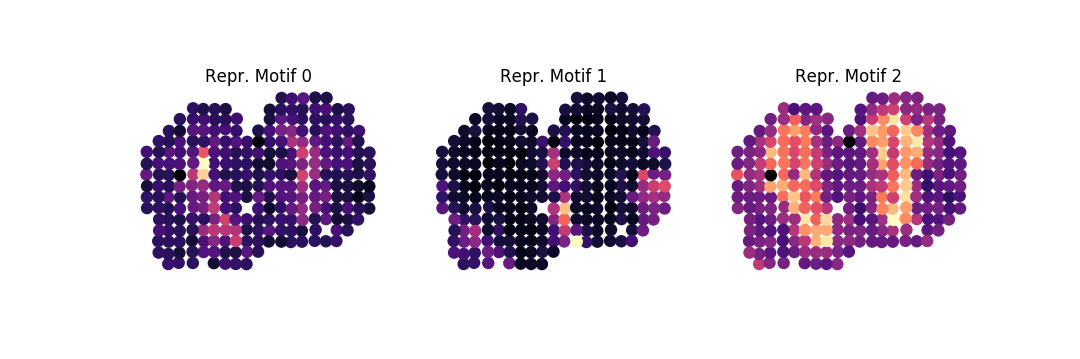
بعد ذلك، لفرز أفضل 100 جين في مجموعة من عائلات الأنماط، حيث يجب تفسير 85% من التباين في أنماطنا من خلال الأنماط الذاتية، قم بما يلي:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3ومن هذا نحصل على العناصر التمثيلية الثلاثة التالية لكل عائلة:
قد نخضع عائلاتنا لتحليل التخصيب، عن طريق تشغيل:
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "حيث نرى على سبيل المثال أن Family 2 مُثري للعديد من العمليات المتعلقة بوظيفة الخلايا العصبية وتوليدها وتنظيمها:
| عائلة | محلي | اسم | p_value | مصدر | intersection_size | |
|---|---|---|---|---|---|---|
| 2 | 2 | اذهب:0007399 | تطور الجهاز العصبي | 0.00035977 | اذهب: بي بي | 26 |
| 3 | 2 | اذهب:0050773 | تنظيم تطور التغصنات | 0.000835883 | اذهب: بي بي | 8 |
| 4 | 2 | اذهب:0048167 | تنظيم اللدونة متشابك | 0.00196494 | اذهب: بي بي | 8 |
| 5 | 2 | اذهب:0016358 | تطور التغصنات | 0.00217167 | اذهب: بي بي | 9 |
| 6 | 2 | اذهب:0048813 | التشكل التغصنات | 0.00741589 | اذهب: بي بي | 7 |
| 7 | 2 | اذهب:0048814 | تنظيم التشكل التغصنات | 0.00800399 | اذهب: بي بي | 6 |
| 8 | 2 | اذهب:0048666 | تطور الخلايا العصبية | 0.0114088 | اذهب: بي بي | 16 |
| 9 | 2 | اذهب:0099004 | مسار إشارات الكيناز المعتمد على الهدوديولين | 0.0159572 | اذهب: بي بي | 3 |
| 10 | 2 | اذهب:0050804 | تعديل انتقال متشابك الكيميائية | 0.0341913 | اذهب: بي بي | 10 |
| 11 | 2 | اذهب:0099177 | تنظيم الإشارات عبر المشبكية | 0.0347783 | اذهب: بي بي | 10 |
وبطبيعة الحال، هذا التحليل ليس شاملا بأي حال من الأحوال. بل مثال سريع لإظهار كيفية تشغيل CLI لـ sepal .
بينما تم تصميم sepal كأداة مستقلة، فقد قمنا أيضًا ببنائها لتكون فعالة كحزمة python قياسية يمكن من خلالها استيراد الوظائف واستخدامها في سير عمل متكامل. لإظهار كيفية القيام بذلك، نقدم مثالاً، إعادة إنتاج تحليل سرطان الجلد. ويمكن إضافة المزيد من الأمثلة في وقت لاحق.
يجب أن يكون الإدخال إلى sepal بالتنسيق n_locations x n_genes ، ولكن إذا تم تنظيم بياناتك بالطريقة المعاكسة ( n_genes x n_locations ) فما عليك سوى توفير علامة --transpose عند تشغيل المحاكاة أو التحليل وسيتم الاهتمام بهذا الأمر ل.
نحن ندعم حاليًا تنسيقات .csv و .tsv و .h5ad . بالنسبة للأخيرة، يجب أن يتم تنظيم ملفك وفقًا لهذا التنسيق. نتوقع أن يكون هناك إصدار من فريق scanpy في المستقبل القريب، حيث يتم تقديم تنسيق موحد للبيانات المكانية، ولكن حتى ذلك الحين سنستخدم المعيار المذكور أعلاه.
جميع البيانات الحقيقية التي استخدمناها عامة، ويمكن الوصول إليها من خلال الروابط التالية:
تم إنشاء البيانات الاصطناعية بواسطة:
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py يمكن العثور على جميع النتائج المقدمة في الدراسة في مجلد res ، سواء بالنسبة للبيانات الحقيقية أو الاصطناعية. قمنا بتنظيم النتائج لكل عينة وفقًا لذلك:
res/sample-name/X-diffusion-times.tsv : أوقات الانتشار لجميع الجينات المرتبةanalysis/ : يحتوي على مخرجات التحليل الثانوي

