binnacle
1.0.0
에게
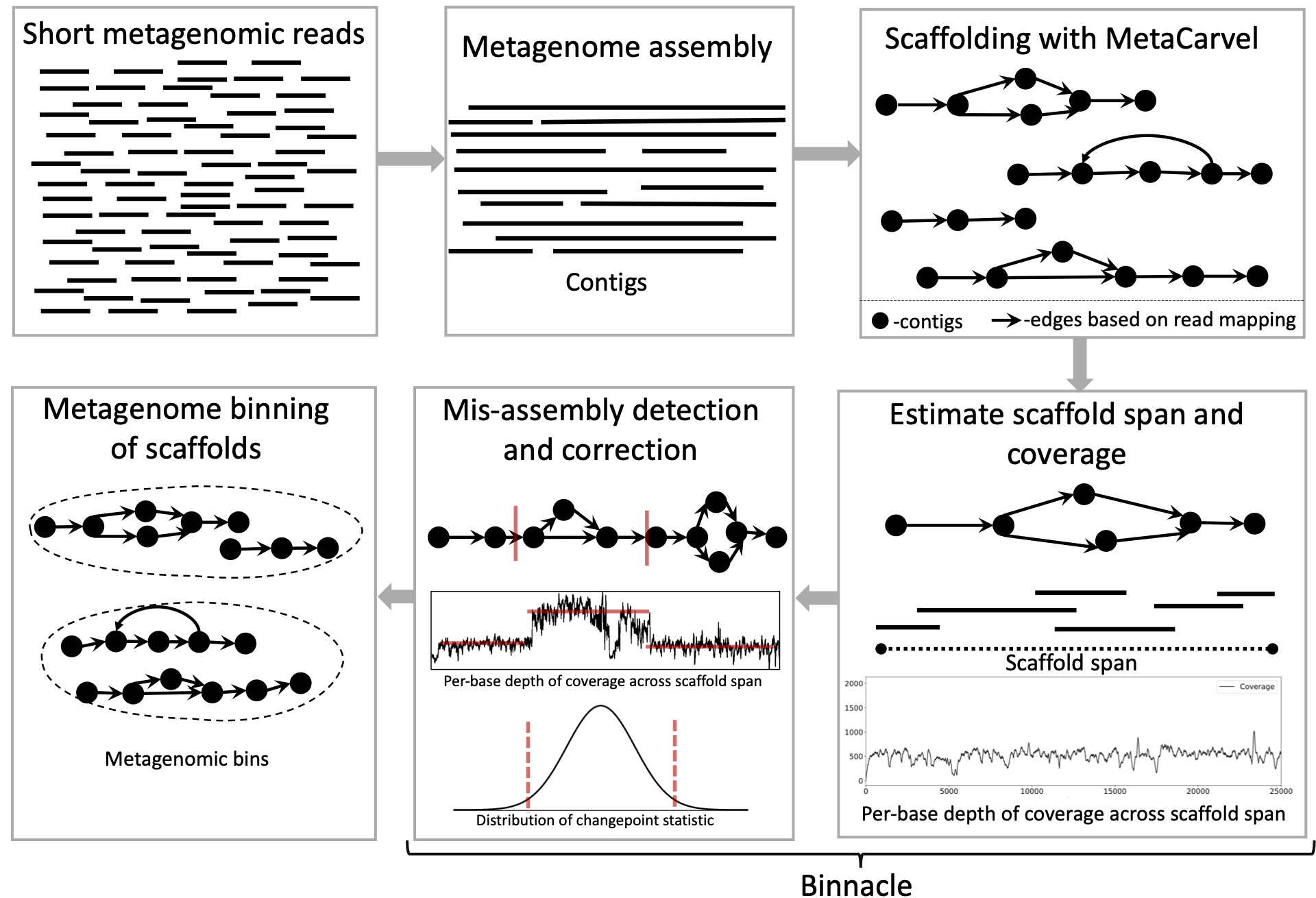
Binnacle은 그래프 스캐 폴드의 적용 범위를 정확하게 계산하고 Metabat2, Maxbin 2.0 및 Concoct와 같은 주요 비닝 방법과 완벽하게 통합됩니다. Binning을위한 Contigs (가장 일반적인 접근법)와 달리 그래프 스캐 폴드를 사용하면 메타 게놈 빈의 연속성과 품질이 향상되고 재구성 된 게놈의 더 넓은 액세서리 요소 세트를 포착 할 수 있습니다.
Binnacle을 실행하려면 Python 3.7.x, BedTools, Samtools, Biopython, NetworkX, Numpy 및 Pandas가 필요합니다.
Environment.yml 파일을 사용할 수 있으며 이는 Binnacle을 실행하는 데 적합한 콘다 환경을 만드는 데 사용될 수 있습니다. 이 패키지를 설치하는 방법에 대한 자세한 설명서는 여기에 제공됩니다. 우리는 Metacarvel 스캐 폴딩 도구의 출력 인 그래프 스캐 폴드를 사용하므로 Metacarvel을 다운로드하여 설치해야합니다. Metacarvel에 대한 단계별 설치 안내서가 있습니다.
일반적으로, 하나 또는 다수의 메타 게놈 샘플이있을 때, 우리는 메타 게놈 빈을 생성하기 위해 각 샘플에서 조립, 스캐 폴드 및 빈 콘티그/스캐 폴드를 조립해야합니다. 어셈블리에는 Megahit을 사용하고 스캐 폴딩에는 Metacarvel을 사용하는 것이 좋습니다. 우리는 조립, 스캐 폴딩 및 기반마다 적용 범위 추정 단계를 통해 작업 할 수있는 도우미 안내서를 제공합니다.
다음 단계에 따라 그래프 스캐 폴드로 비닝 방법을 실행하기위한 파일을 생성하십시오.
python Estimate_Abundances.py -g [ORIENTED.gml] -a [COVERAGE_SORTED.txt] -c [CONTIGS.fa] -d [OUTPUT_DIRECTORY]
usage: Estimate_Abundances.py [-h] [-g ASSEMBLY] [-a COVERAGE] [-bam BAMFILE]
[-bed BEDFILE] [-c CONTIGS] -d DIR [-o COORDS]
[-w WINDOW_SIZE] [-t THRESHOLD]
[-n NEIGHBOR_CUTOFF] [-p POSCUTOFF]
[-pre PREFIX]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel. Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. If the coordinates computed by binnacle
is specified then the abundance for each scaffold is estimated based on the
contig abundances and the coordinates. If the coordinates are not specified
then binnacle etimates the abundance from scratch. While calculating all vs
all abundances please specify the coordinates(Coordinates_After_Delinking.txt)
through the "coords" parameter. The abundances can be provided as a bed file,
bam file or a text file describing the per base coverage obtained by running
the genomeCoverageBed program of the bedtools suite.
optional arguments:
-h, --help show this help message and exit
-g ASSEMBLY, --assembly ASSEMBLY
Assembly Graph generated by Metacarvel
-a COVERAGE, --coverage COVERAGE
Output generated by running genomecov -d on the bed
file generated by MetaCarvel.
-bam BAMFILE, --bamfile BAMFILE
Bam file from aligning reads to contigs
-bed BEDFILE, --bedfile BEDFILE
Bed file from aligning reads to contigs. If bed file
is provided please provide a fasta file of the contigs
-c CONTIGS, --contigs CONTIGS
Contigs generated by the assembler, contigs.fasta
-d DIR, --dir DIR output directory for results
-o COORDS, --coords COORDS
Coordinate file generated by Binnacle
-w WINDOW_SIZE, --window_size WINDOW_SIZE
Size of the sliding window for computing test
statistic to identify changepoints in coverages
(Default=1500)
-t THRESHOLD, --threshold THRESHOLD
Threshold to identify outliers (Default=99)
-n NEIGHBOR_CUTOFF, --neighbor_cutoff NEIGHBOR_CUTOFF
Filter size to identify outliers within (Defualt=100)
-p POSCUTOFF, --poscutoff POSCUTOFF
Position cutoff to consider delinking (Default=100)
-pre PREFIX, --prefix PREFIX
Prefix to be attached to all outputs
-g Path to oriented.gml from running metacarvel on sample
-c Path to contigs obtained by assembling reads of sample
-a Coverage of contigs in ths sample by mapping to its reads -- See Wiki for how to calculate coverage information
-d Output directory
-a Coverage of contigs in Sample 1 by mapping reads of Sample 2 -- See Wiki for how to calculate coverage information
-o Coordinates of scaffolds from Sample 1 that you would have generated from the previous step.
-d Same output directory as Sample 1
python Collate.py -h
usage: Collate.py [-h] -d DIR [-m METHOD] [-k KEEP]
binnacle: A tool for binning metagenomic datasets using assembly graphs and
scaffolds generated by metacarvel.Estimate_Abundances.py estimates abundance
for scaffolds generated by MetaCarvel. The program Collate.py collects the
summary files generated by Estimate_Abundances.py
optional arguments:
-h, --help show this help message and exit
-d DIR, --dir DIR Output directory that contains the summary files
generated by running Estimate_Abundances.py
-m METHOD, --method METHOD
Binning method to format the output to. Presently we
support 1. Metabat 2. Maxbin 3. Concoct 4. Binnacle
(Default)
-k KEEP, --keep KEEP Retain the summary files generated by
Estimate_Abundances.py. Defaults to True
파이썬 환경 설정, 적용 범위를 계산하는 방법 및 Binnacle을 실행하기위한 일반적인 워크 플로우에 대한 자세한 설명은 위키를 확인하십시오.
그래프 스캐 폴드를 시각화하려면 웹 기반 브라우저 인 Metagenomescope를 사용하는 것이 좋습니다. Metagenomescope에 대한 입력은 astembly_graph_filtered.gml입니다. Metagenomescope 설치 및 실행에 대한 자세한 문서는 여기에 제공됩니다.
Muralidharan HS, Shah N, Meisel JS 및 Pop M (2021) Binnacle : 스캐 폴드를 사용하여 메타 게놈 빈의 연속성과 품질을 향상시킵니다. 앞쪽. 미생물. 12 : 638561. doi : 10.3389/fmicb.2021.638561.
이 도구는 여전히 개발 중입니다. 궁금한 점이 있으면 여기 GitHub에서 문제를 열거나 당사에 문의하십시오.
Harihara Muralidharan : [email protected]
nidhi shah : [email protected]


