이 저장소에는 다음이 포함되어 있습니다.
sepal python3 필요하며, 가급적이면 3.5 이상의 버전이 필요합니다. 다운로드 및 설치하려면 터미널을 열고 sepal 다운로드할 디렉터리로 변경한 후 다음을 수행하세요.
git clone https://github.com/almaan/sepal.git
cd sepal
chmod +x setup.py
./setup.py install
사용자 권한에 따라 --user setup.py 에 인수로 추가해야 할 수도 있습니다. 설정을 실행하면 확산 시간을 계산하는 데 필요한 최소한의 설치가 제공됩니다. 그러나 분석 모듈을 사용하려면 권장 패키지도 설치해야 합니다. 이렇게 하려면 (동일한 디렉터리에서) 다음을 실행하면 됩니다.
pip install -e " .[full] " 다시 말하지만 --user 포함해야 할 수도 있습니다. 또한 이것이 python-pip 인터페이스를 설정한 방식인 경우 pip3 사용해야 할 수도 있습니다. conda 또는 가상 환경을 사용하는 경우 패키지 설치에 대한 권장 사항을 따르십시오.
이렇게 하면 명령줄 인터페이스(CLI)와 표준 패키지가 모두 설치되어야 합니다. 설치가 성공했는지 테스트하고 확인하려면 다음 명령을 실행해 볼 수 있습니다.
sepal -h
sepal과 관련된 도움말 메시지를 인쇄해야 합니다. 지금까지 모든 것이 잘 진행되었다면 예제 섹션으로 이동하여 sepal 실제로 작동하는 모습을 볼 수 있습니다!
sepal의 권장 사용은 명령줄 인터페이스를 사용하는 것입니다. 확산 시간을 계산하기 위한 시뮬레이션과 결과의 후속 분석 또는 검사는 sepal 입력하고 run 또는 analyze 수행하여 쉽게 수행할 수 있습니다. analyze 모듈에는 결과 시각화( inspect ), 프로필을 패턴 계열( family )로 정렬 또는 식별된 계열에 기능 강화 분석( fea )을 적용하는 등 다양한 옵션이 있습니다. 사용 가능한 전체 명령 목록을 보려면 sepal module -h 수행하세요. 여기서 module 은 run 및 analyze 중 하나입니다. 아래에서는 sepal을 사용하여 공간 패턴이 있는 전사 프로필을 찾는 방법을 보여줍니다.
결과를 저장할 폴더를 생성할 것입니다. 이 폴더는 작업 디렉터리로도 사용됩니다. 저장소의 기본 디렉터리에서 다음을 수행합니다.
cd res
mkdir example
cd exampleMOB 샘플은 분석의 예시로 사용됩니다. 각 전사 프로필에 대한 확산 시간 계산부터 시작합니다.
sepal run -c ../../data/real/mob.tsv.gz -mo 10 -mc 5 -o . -ar 1다음은 이것이 어떻게 보일지에 대한 예(help 명령의 추가 표시 포함)입니다.

확산 시간을 계산한 후 결과를 조사하고 싶습니다. 연구에서처럼 상위 20개 프로필을 살펴보겠습니다. 다음 명령을 실행하여 결과에서 이미지를 쉽게 생성할 수 있습니다.
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . inspect -ng 20 -nc 5다음과 같은 내용이 보일 것입니다.

출력은 다음 이미지가 됩니다.
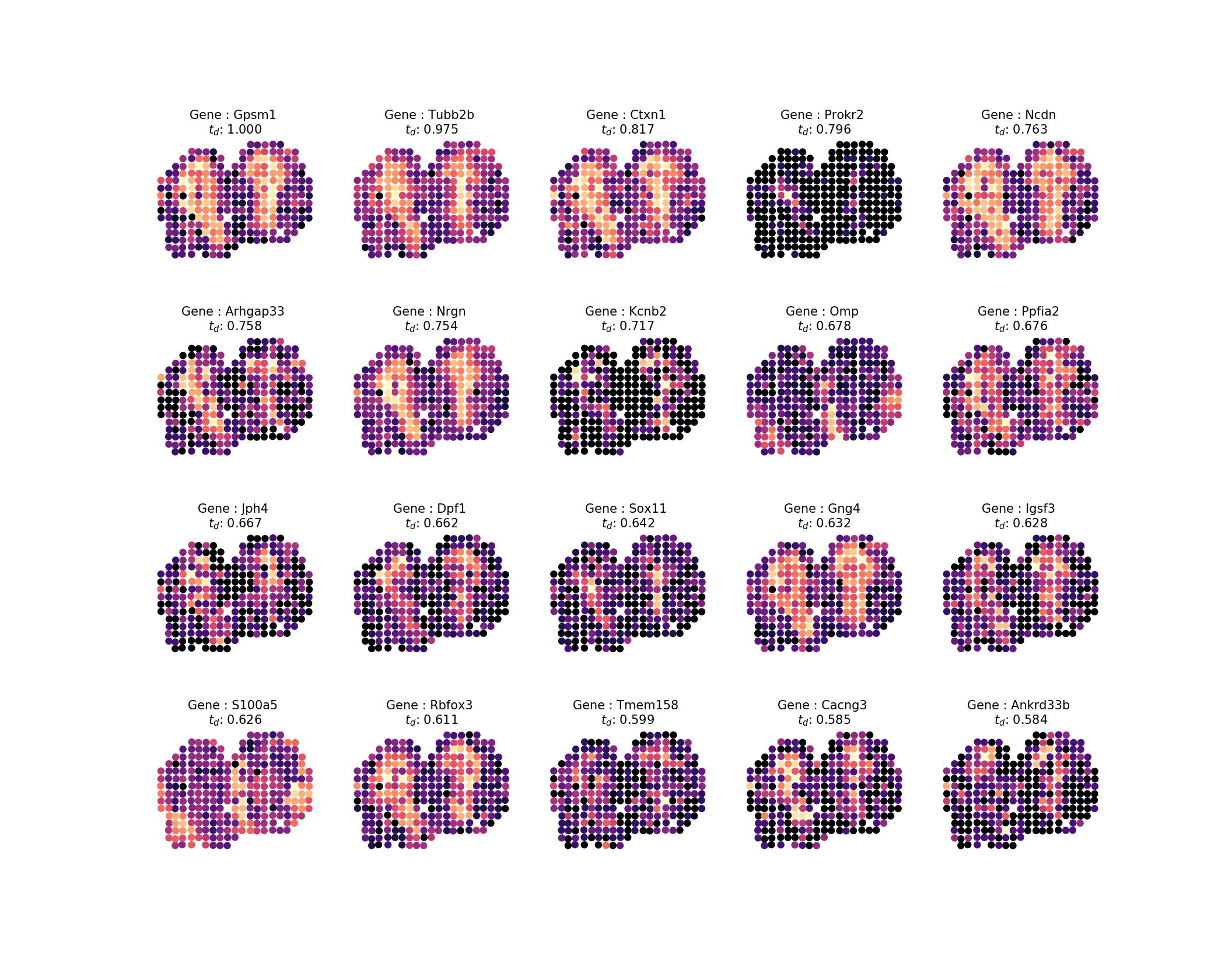
그런 다음 상위 100개 유전자를 패턴군 세트로 정렬하려면 다음을 수행하십시오. 여기서 패턴 분산의 85%는 고유 패턴으로 설명됩니다.
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . family -ng 100 -nbg 100 -eps 0.85 --plot -nc 3이를 통해 우리는 각 가문에 대한 다음과 같은 세 가지 대표적인 모티프를 얻습니다.
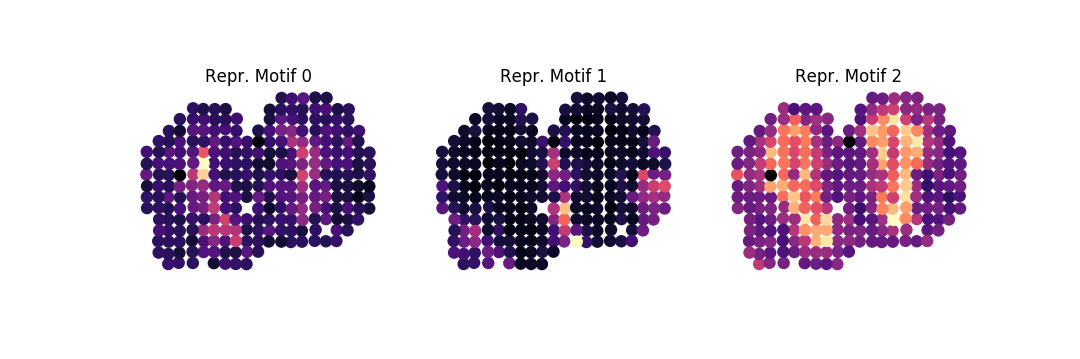
다음을 실행하여 가족을 강화 분석할 수 있습니다.
sepal analyze -c ../../data/real/mob.tsv.gz
-r 20200409173043610345-top-diffusion-times.tsv
-ar 1k -o . fea -fl mob.tsv-family-index.tsv -or " mmusculus "예를 들어 Family 2에는 신경 기능, 생성 및 조절과 관련된 여러 프로세스가 풍부하다는 것을 알 수 있습니다.
| 가족 | 토종의 | 이름 | p_값 | 원천 | 교차로_크기 | |
|---|---|---|---|---|---|---|
| 2 | 2 | GO:0007399 | 신경계 발달 | 0.00035977 | GO:BP | 26 |
| 3 | 2 | GO:0050773 | 수상돌기 발달 조절 | 0.000835883 | GO:BP | 8 |
| 4 | 2 | GO:0048167 | 시냅스 가소성 조절 | 0.00196494 | GO:BP | 8 |
| 5 | 2 | GO:0016358 | 수상돌기 발달 | 0.00217167 | GO:BP | 9 |
| 6 | 2 | GO:0048813 | 수상돌기 형태형성 | 0.00741589 | GO:BP | 7 |
| 7 | 2 | GO:0048814 | 수상돌기 형태형성의 조절 | 0.00800399 | GO:BP | 6 |
| 8 | 2 | GO:0048666 | 뉴런 발달 | 0.0114088 | GO:BP | 16 |
| 9 | 2 | GO:0099004 | 칼모듈린 의존성 키나제 신호전달 경로 | 0.0159572 | GO:BP | 3 |
| 10 | 2 | GO:0050804 | 화학적 시냅스 전달의 조절 | 0.0341913 | GO:BP | 10 |
| 11 | 2 | GO:0099177 | 시냅스 신호의 조절 | 0.0347783 | GO:BP | 10 |
물론 이 분석이 결코 완전한 것은 아니다. 그러나 sepal 에 대해 CLI를 작동하는 방법을 보여주는 간단한 예입니다.
sepal 독립 실행형 도구로 설계되었지만 통합 워크플로에서 기능을 가져와 사용할 수 있는 표준 Python 패키지로도 작동하도록 구성했습니다. 이것이 어떻게 수행될 수 있는지 보여주기 위해 흑색종 분석을 재현하는 예를 제공합니다. 나중에 더 많은 예제가 추가될 수 있습니다.
sepal 에 대한 입력은 n_locations x n_genes 형식이어야 합니다. 그러나 데이터가 반대 방식( n_genes x n_locations )으로 구성된 경우 시뮬레이션이나 분석을 실행할 때 --transpose 플래그를 제공하면 이 문제가 처리됩니다. 의.
현재 .csv , .tsv 및 .h5ad 형식을 지원합니다. 후자의 경우 파일은 이 형식에 따라 구성되어야 합니다. 가까운 시일 내에 공간 데이터에 대한 표준화된 형식이 제시되는 scanpy 팀의 릴리스가 있을 것으로 예상하지만 그때까지는 앞서 언급한 표준을 사용할 것입니다.
우리가 사용한 모든 실제 데이터는 공개되어 있으며 다음 링크에서 액세스할 수 있습니다.
합성 데이터는 다음에 의해 생성되었습니다.
synthetic/img2cnt.pysynthetic/turing.pysynthetic/ablation.py 연구에 제시된 모든 결과는 실제 데이터와 합성 데이터 모두 res 폴더에서 찾을 수 있습니다. 각 샘플에 대해 그에 따라 결과를 구성했습니다.
res/sample-name/X-diffusion-times.tsv : 순위가 매겨진 모든 유전자의 확산 시간analysis/ : 2차 분석 결과를 포함합니다.

